

Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutations
- PMID: 26195756
- PMCID: PMC4517250
- DOI: 10.1073/pnas.1501713112
Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutations
Abstract
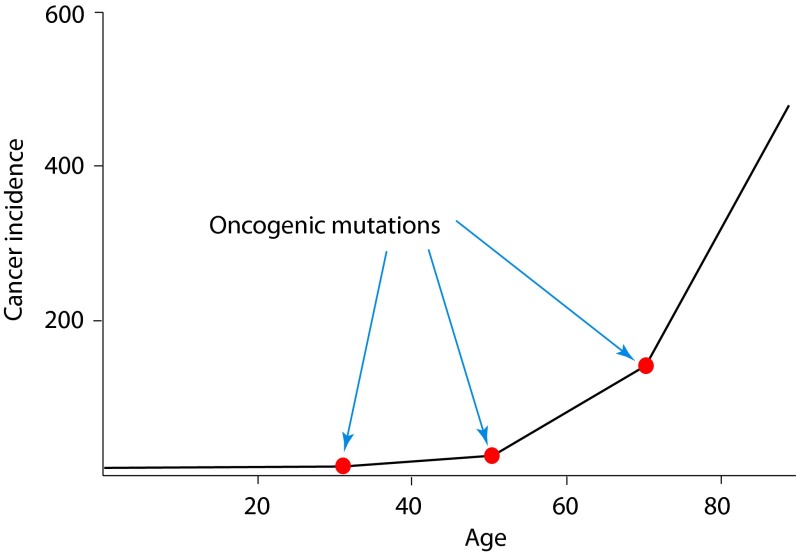
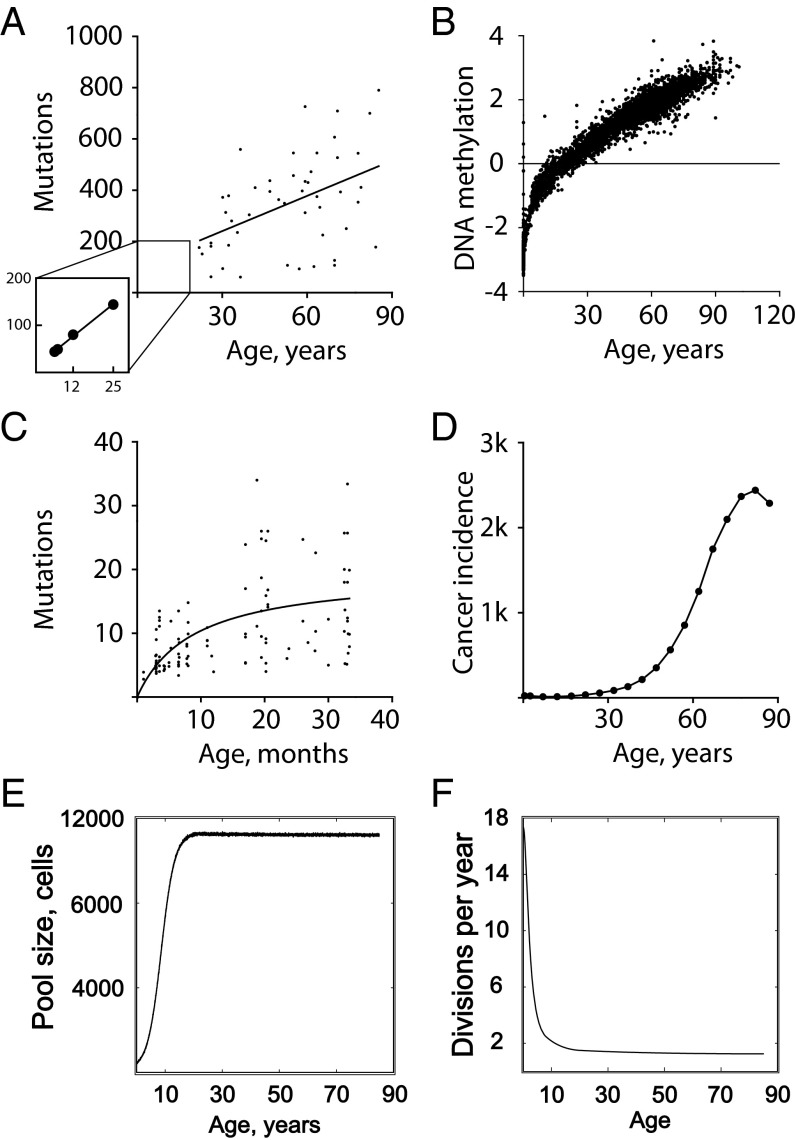
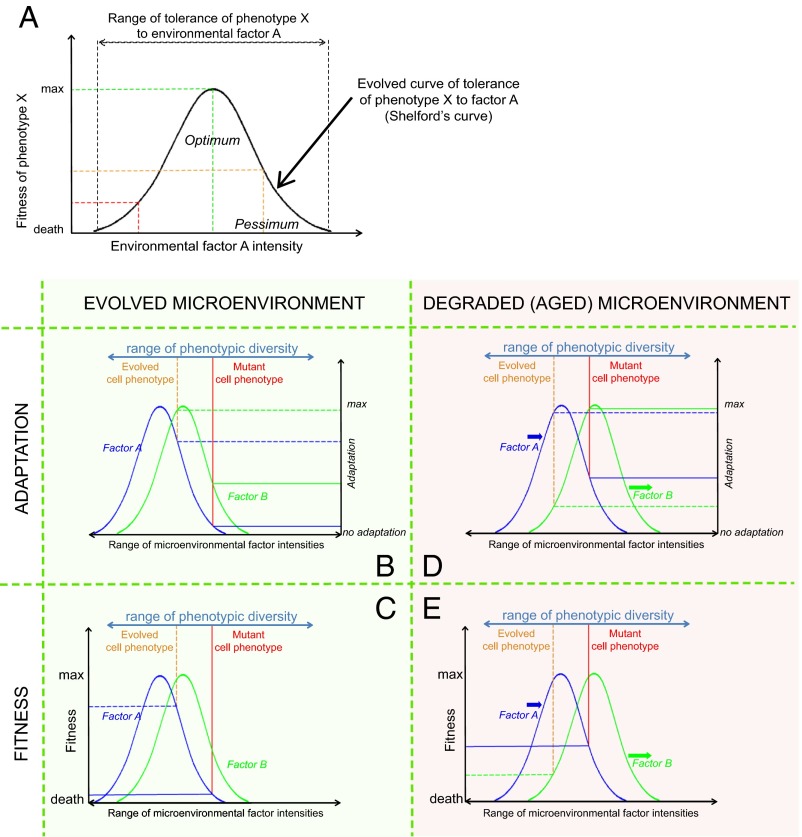
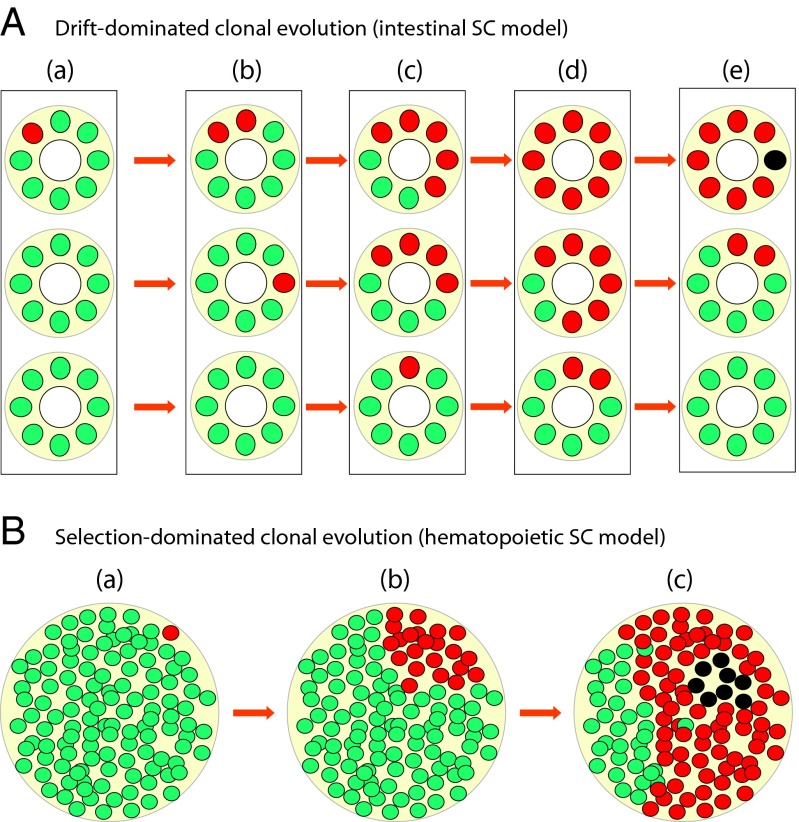
Our understanding of cancer has greatly advanced since Nordling [Nordling CO (1953) Br J Cancer 7(1):68-72] and Armitage and Doll [Armitage P, Doll R (1954) Br J Cancer 8(1):1-12] put forth the multistage model of carcinogenesis. However, a number of observations remain poorly understood from the standpoint of this paradigm in its contemporary state. These observations include the similar age-dependent exponential rise in incidence of cancers originating from stem/progenitor pools differing drastically in size, age-dependent cell division profiles, and compartmentalization. This common incidence pattern is characteristic of cancers requiring different numbers of oncogenic mutations, and it scales to very divergent life spans of mammalian species. Also, bigger mammals with larger underlying stem cell pools are not proportionally more prone to cancer, an observation known as Peto's paradox. Here, we present a number of factors beyond the occurrence of oncogenic mutations that are unaccounted for in the current model of cancer development but should have significant impacts on cancer incidence. Furthermore, we propose a revision of the current understanding for how oncogenic and other functional somatic mutations affect cellular fitness. We present evidence, substantiated by evolutionary theory, demonstrating that fitness is a dynamic environment-dependent property of a phenotype and that oncogenic mutations should have vastly different fitness effects on somatic cells dependent on the tissue microenvironment in an age-dependent manner. Combined, this evidence provides a firm basis for understanding the age-dependent incidence of cancers as driven by age-altered systemic processes regulated above the cell level.
Keywords: aging; cancer; fitness; oncogenic mutations; somatic evolution.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
