

Identification of a Set of Conserved Eukaryotic Internal Retention Time Standards for Data-independent Acquisition Mass Spectrometry
- PMID: 26199342
- PMCID: PMC4597153
- DOI: 10.1074/mcp.O114.042267
Identification of a Set of Conserved Eukaryotic Internal Retention Time Standards for Data-independent Acquisition Mass Spectrometry
Abstract
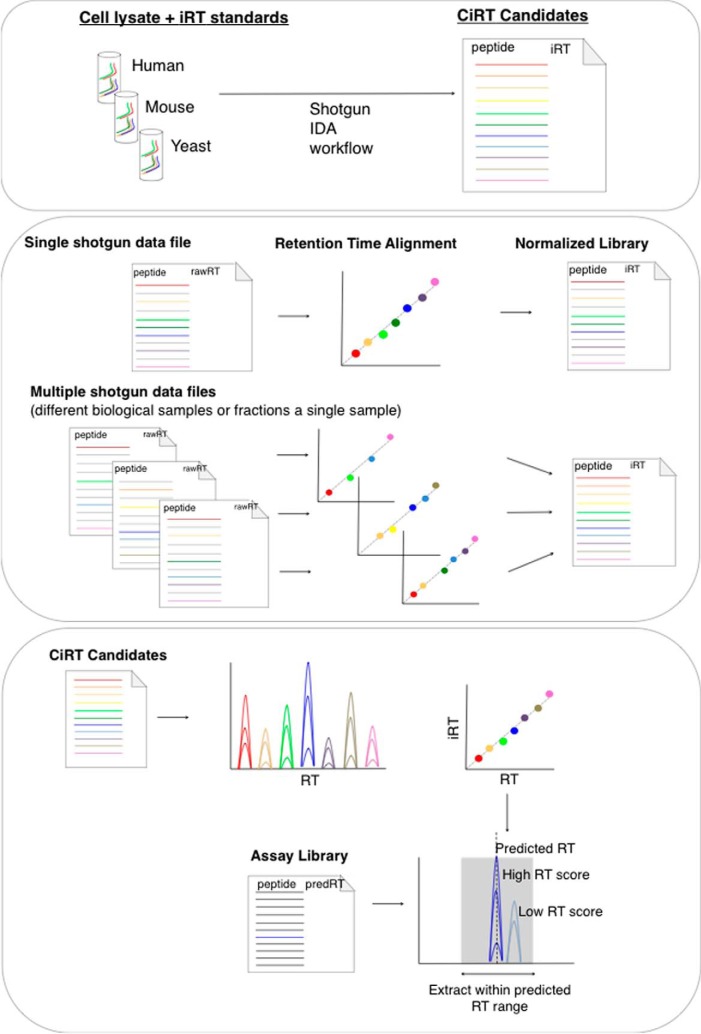
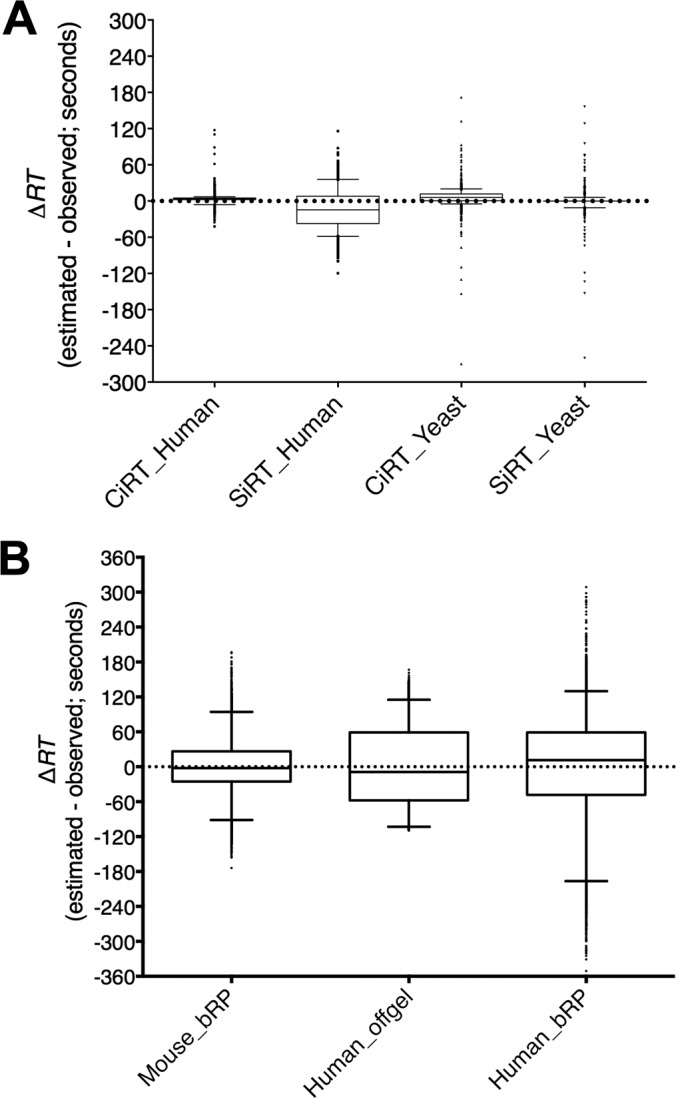
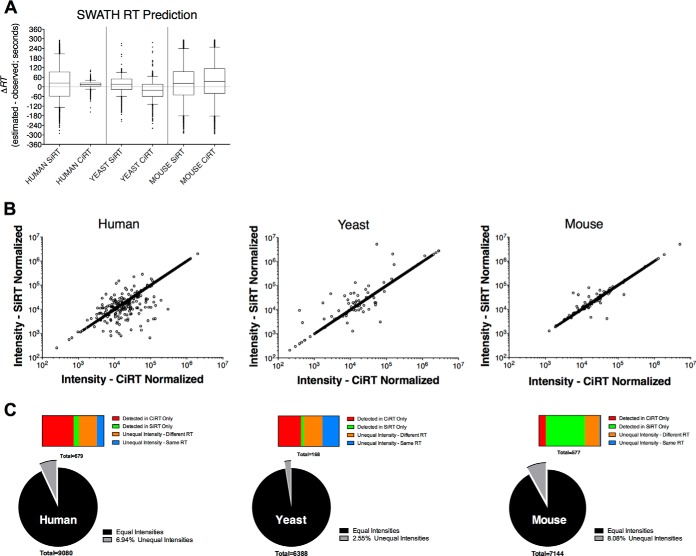
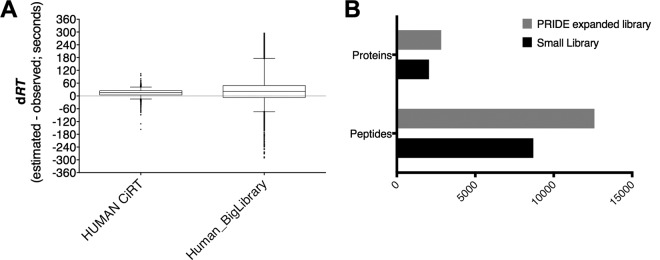
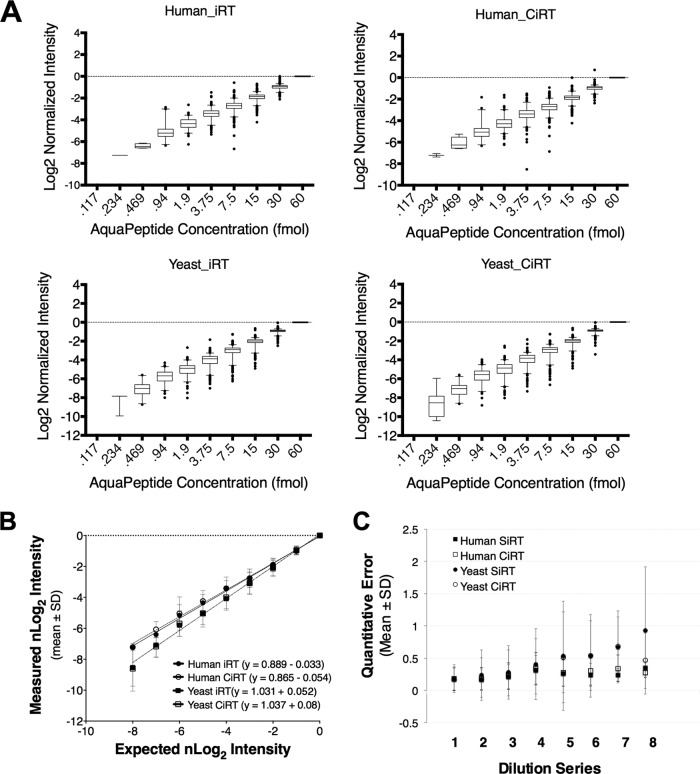
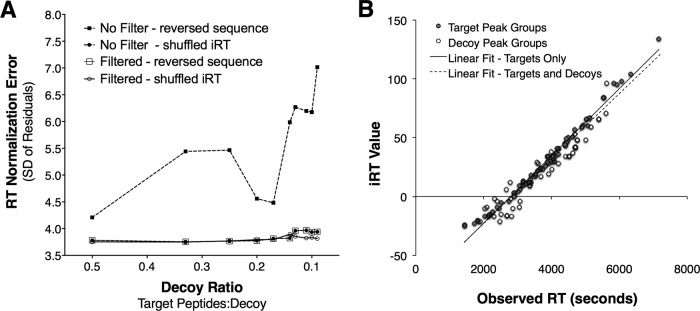
Accurate knowledge of retention time (RT) in liquid chromatography-based mass spectrometry data facilitates peptide identification, quantification, and multiplexing in targeted and discovery-based workflows. Retention time prediction is particularly important for peptide analysis in emerging data-independent acquisition (DIA) experiments such as SWATH-MS. The indexed RT approach, iRT, uses synthetic spiked-in peptide standards (SiRT) to set RT to a unit-less scale, allowing for normalization of peptide RT between different samples and chromatographic set-ups. The obligatory use of SiRTs can be costly and complicates comparisons and data integration if standards are not included in every sample. Reliance on SiRTs also prevents the inclusion of archived mass spectrometry data for generation of the peptide assay libraries central to targeted DIA-MS data analysis. We have identified a set of peptide sequences that are conserved across most eukaryotic species, termed Common internal Retention Time standards (CiRT). In a series of tests to support the appropriateness of the CiRT-based method, we show: (1) the CiRT peptides normalized RT in human, yeast, and mouse cell lysate derived peptide assay libraries and enabled merging of archived libraries for expanded DIA-MS quantitative applications; (2) CiRTs predicted RT in SWATH-MS data within a 2-min margin of error for the majority of peptides; and (3) normalization of RT using the CiRT peptides enabled the accurate SWATH-MS-based quantification of 340 synthetic isotopically labeled peptides that were spiked into either human or yeast cell lysate. To automate and facilitate the use of these CiRT peptide lists or other custom user-defined internal RT reference peptides in DIA workflows, an algorithm was designed to automatically select a high-quality subset of datapoints for robust linear alignment of RT for use. Implementations of this algorithm are available for the OpenSWATH and Skyline platforms. Thus, CiRT peptides can be used alone or as a complement to SiRTs for RT normalization across peptide spectral libraries and in quantitative DIA-MS studies.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Klammer A. A., Yi X., MacCoss M. J., Noble W. S. (2007) Improving tandem mass spectrum identification using peptide retention time prediction across diverse chromatography conditions. Anal. Chem. 79, 6111–6118 - PubMed
-
- Pfeifer N., Leinenbach A., Huber C. G., Kohlbacher O. (2009) Improving peptide identification in proteome analysis by a two-dimensional retention time filtering approach. J. Proteome Res. 8, 4109–4115 - PubMed
-
- Gallien S., Peterman S., Kiyonami R., Souady J., Duriez E., Schoen A., Domon B. (2012) Highly multiplexed targeted proteomics using precise control of peptide retention time. Proteomics 12, 1122–1133 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
