

Viral dark matter and virus-host interactions resolved from publicly available microbial genomes
- PMID: 26200428
- PMCID: PMC4533152
- DOI: 10.7554/eLife.08490
Viral dark matter and virus-host interactions resolved from publicly available microbial genomes
Abstract
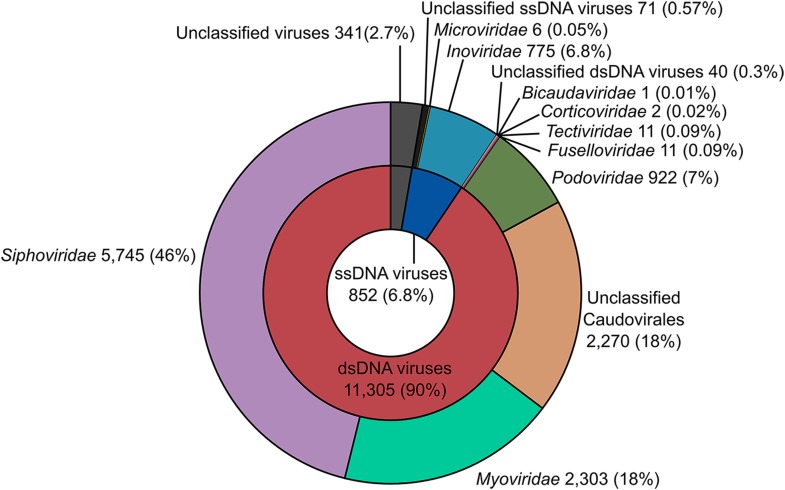
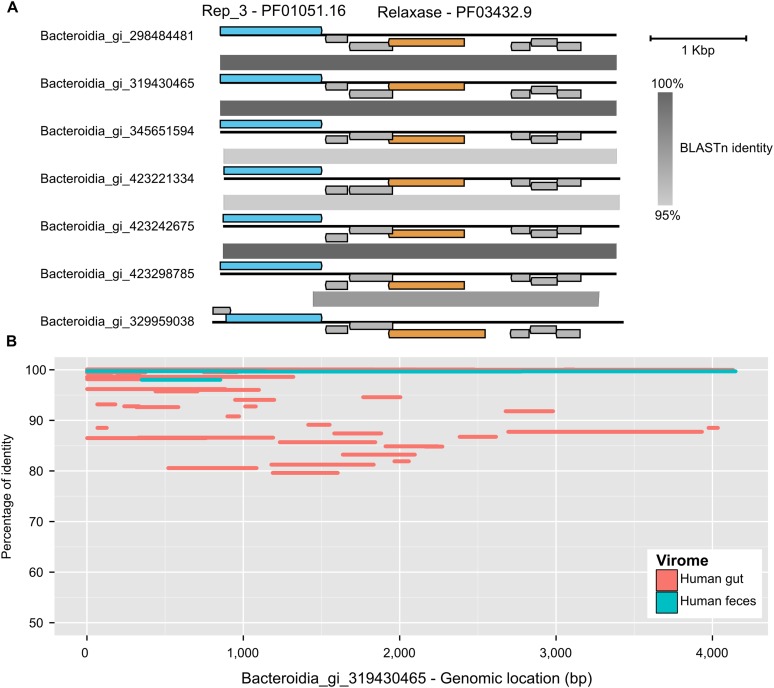
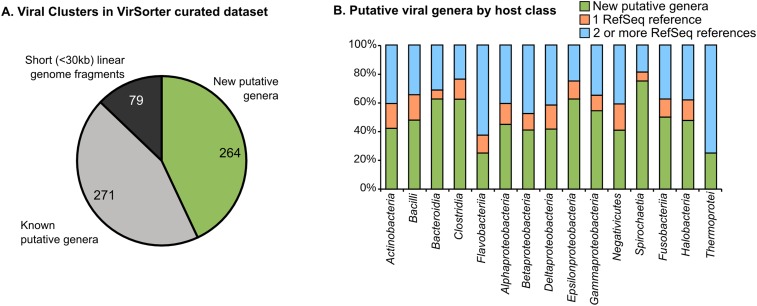
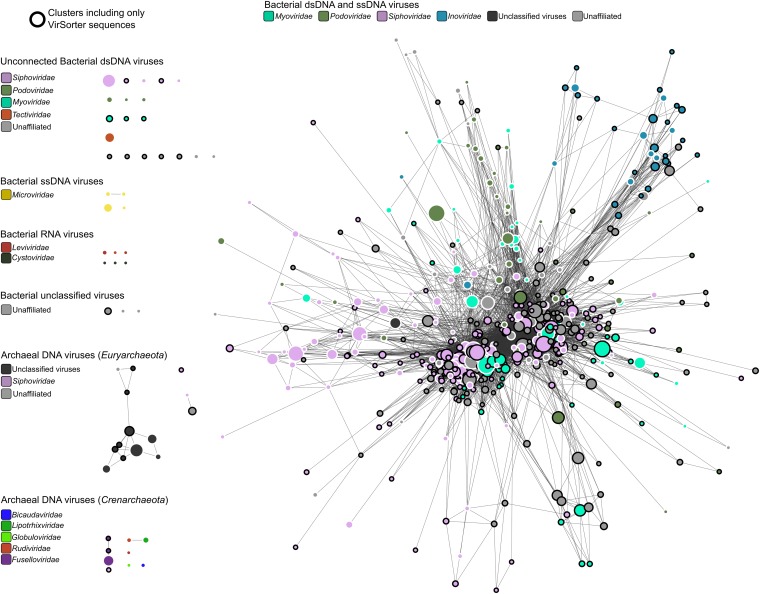
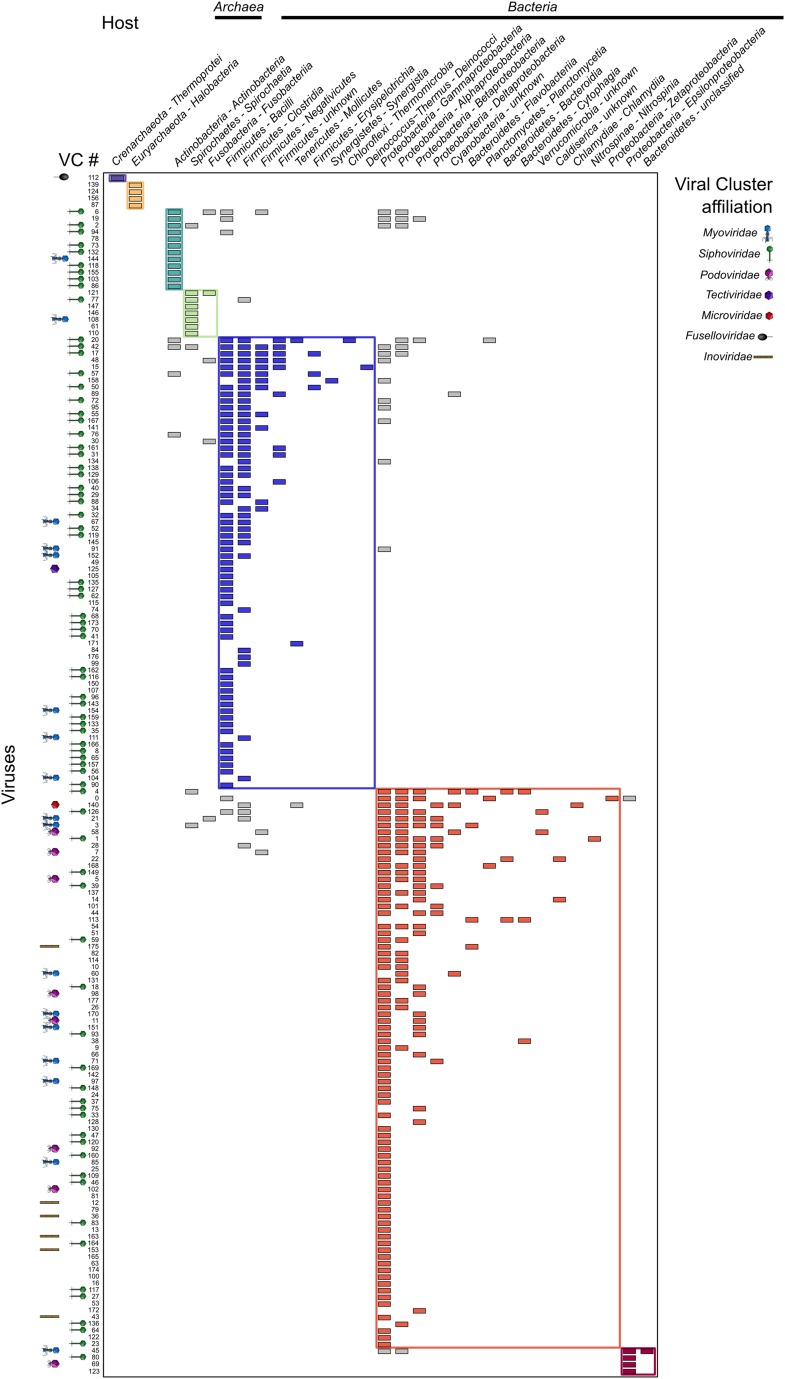
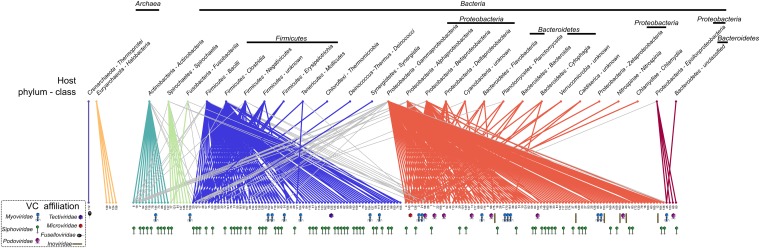
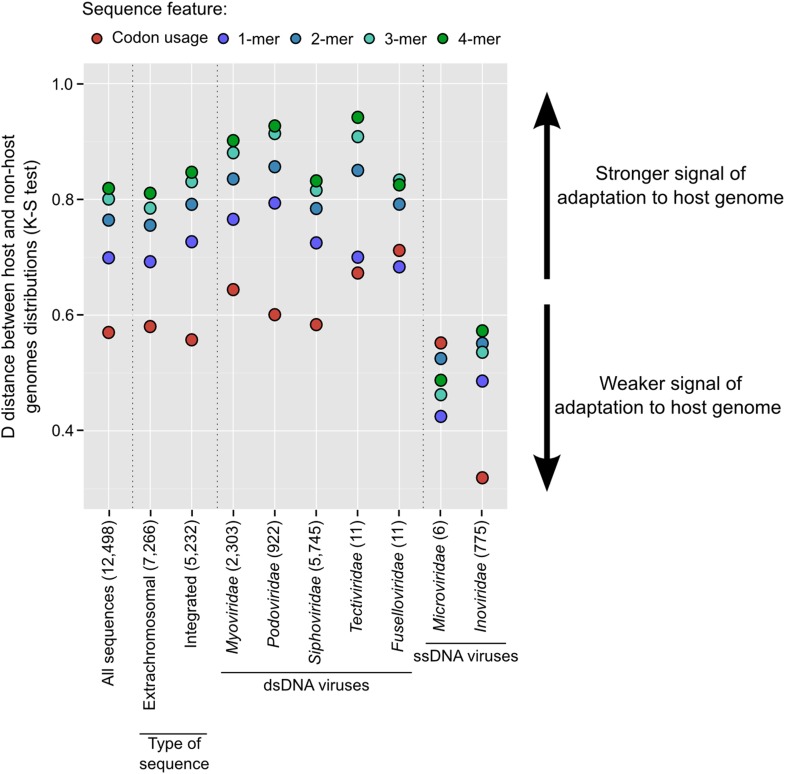
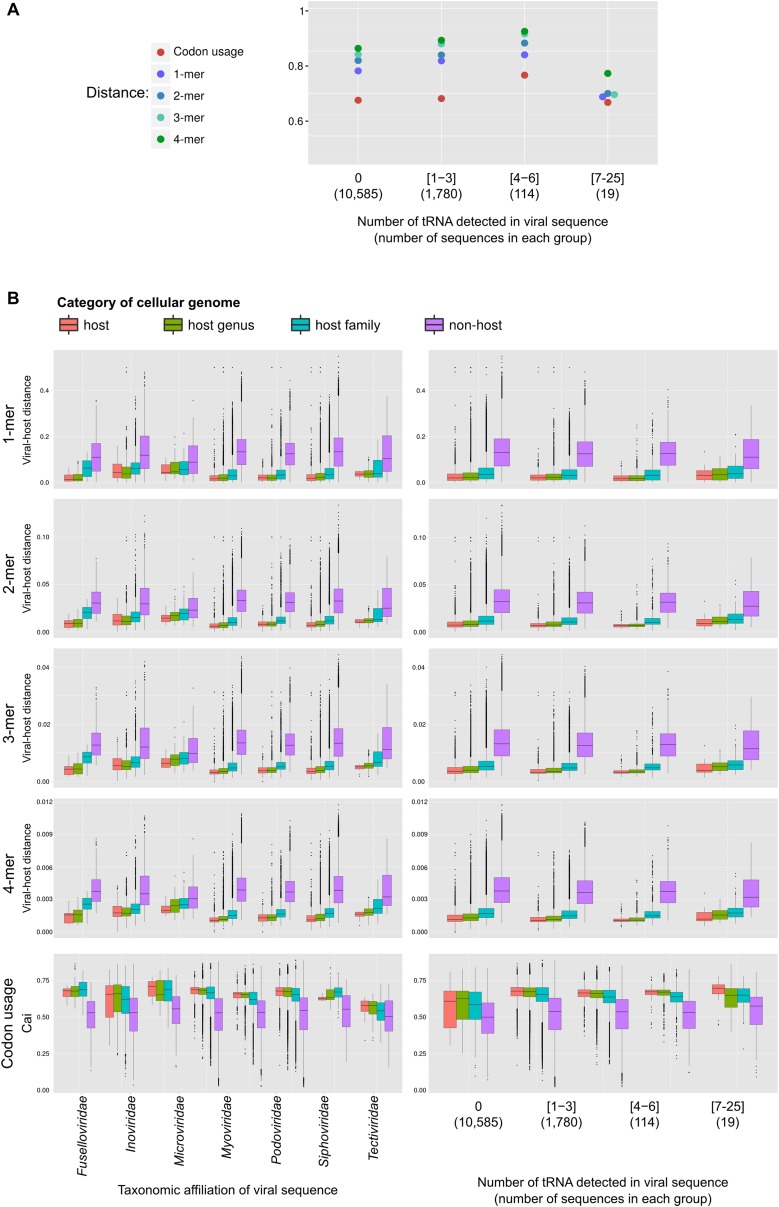
The ecological importance of viruses is now widely recognized, yet our limited knowledge of viral sequence space and virus-host interactions precludes accurate prediction of their roles and impacts. In this study, we mined publicly available bacterial and archaeal genomic data sets to identify 12,498 high-confidence viral genomes linked to their microbial hosts. These data augment public data sets 10-fold, provide first viral sequences for 13 new bacterial phyla including ecologically abundant phyla, and help taxonomically identify 7-38% of 'unknown' sequence space in viromes. Genome- and network-based classification was largely consistent with accepted viral taxonomy and suggested that (i) 264 new viral genera were identified (doubling known genera) and (ii) cross-taxon genomic recombination is limited. Further analyses provided empirical data on extrachromosomal prophages and coinfection prevalences, as well as evaluation of in silico virus-host linkage predictions. Together these findings illustrate the value of mining viral signal from microbial genomes.
Keywords: ecology; evolutionary biology; genomics; none; phage; prophage; virus; virus-host adaptation.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures

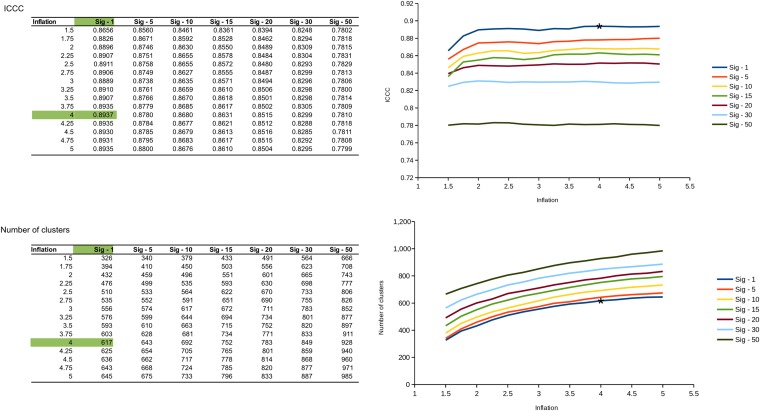
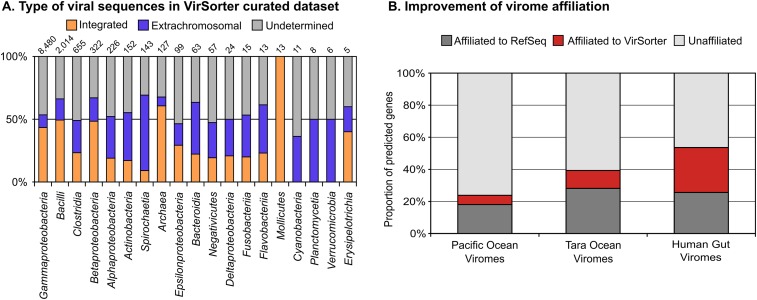
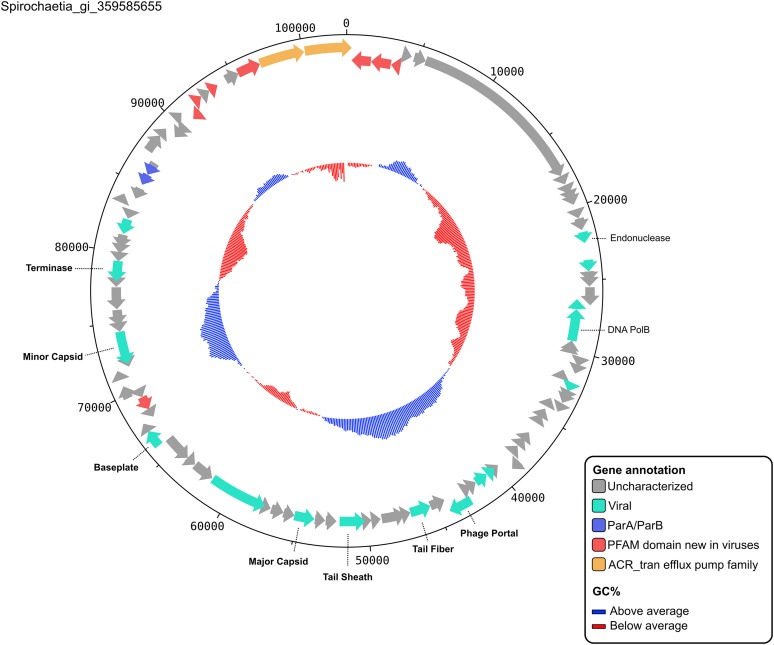
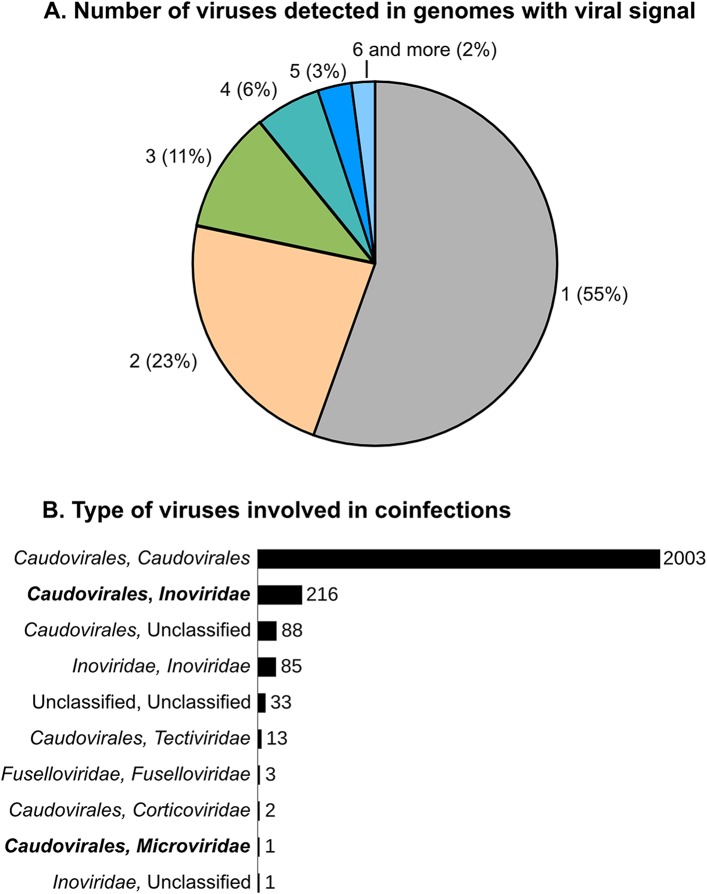
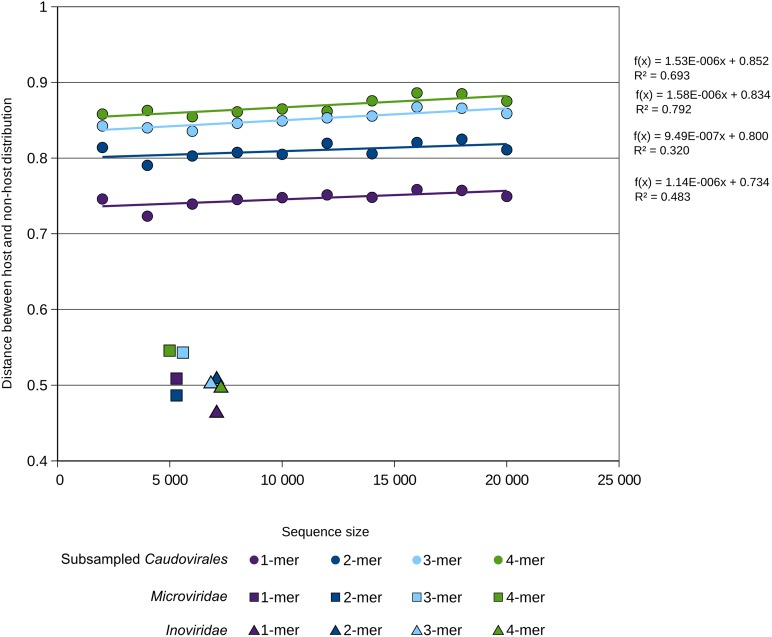
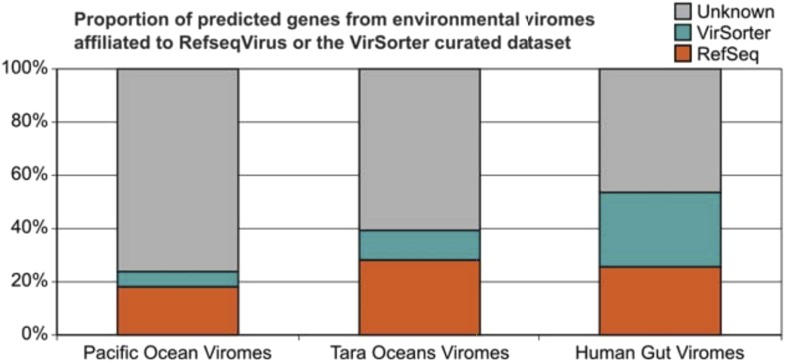
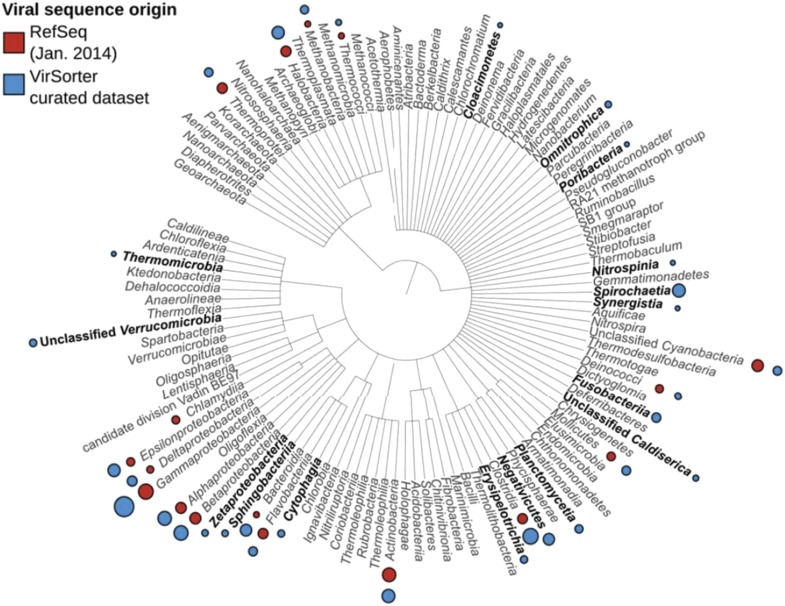
Comment in
-
Microbial ecology: Sorting out viral dark matter.Nat Rev Microbiol. 2015 Sep;13(9):526-7. doi: 10.1038/nrmicro3541. Nat Rev Microbiol. 2015. PMID: 26272401 No abstract available.
References
-
- Allers E, Moraru C, Duhaime MB, Beneze E, Solonenko N, Canosa JB, Amann R, Sullivan MB. Single-cell and population level viral infection dynamics revealed by phageFISH, a method to visualize intracellular and free viruses. Environmental Microbiology. 2013a;15:2306–2318. doi: 10.1111/1462-2920.12100. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
