

Comparative assessment of methods for the computational inference of transcript isoform abundance from RNA-seq data
- PMID: 26201343
- PMCID: PMC4511015
- DOI: 10.1186/s13059-015-0702-5
Comparative assessment of methods for the computational inference of transcript isoform abundance from RNA-seq data
Abstract
Background: Understanding the regulation of gene expression, including transcription start site usage, alternative splicing, and polyadenylation, requires accurate quantification of expression levels down to the level of individual transcript isoforms. To comparatively evaluate the accuracy of the many methods that have been proposed for estimating transcript isoform abundance from RNA sequencing data, we have used both synthetic data as well as an independent experimental method for quantifying the abundance of transcript ends at the genome-wide level.
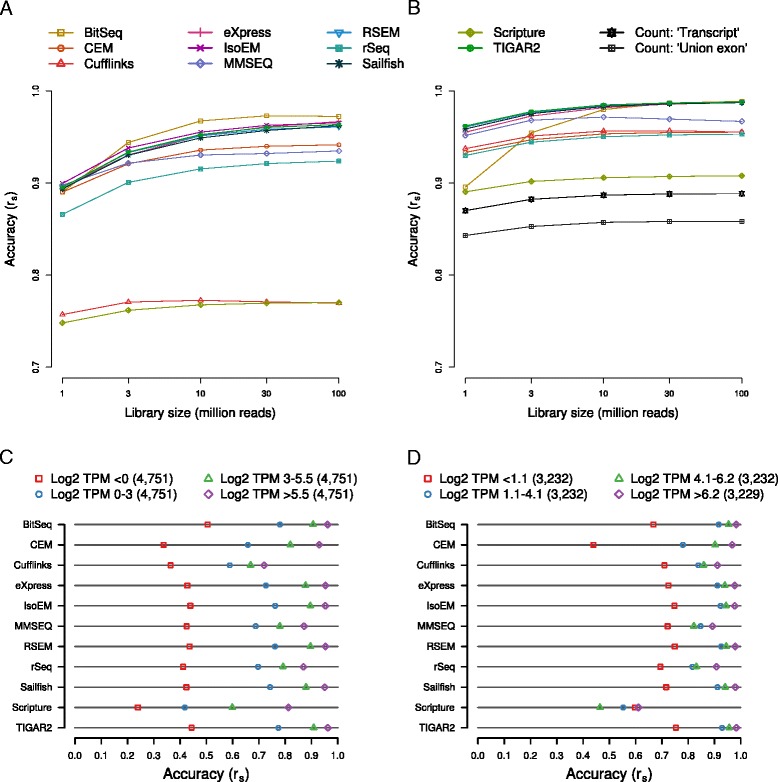
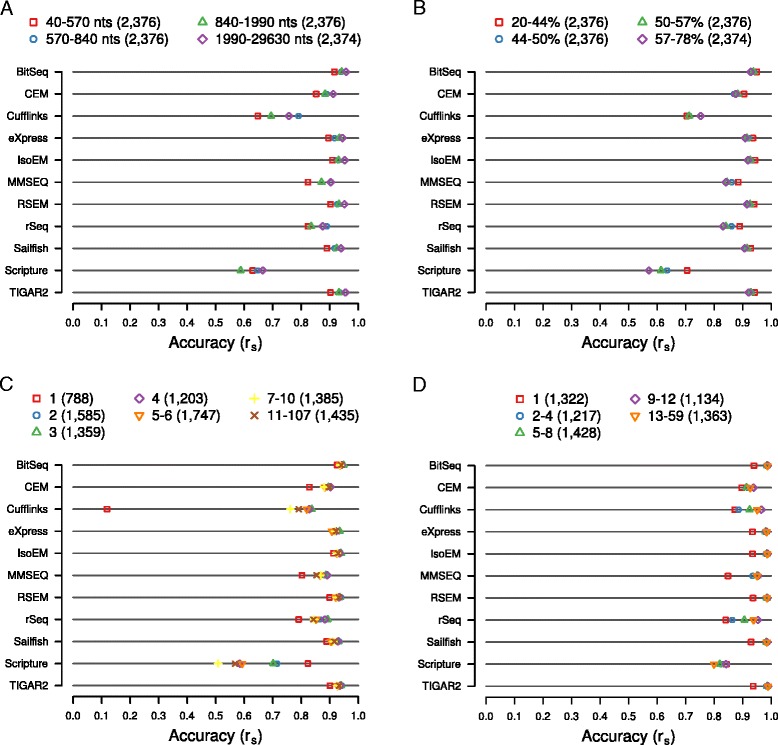
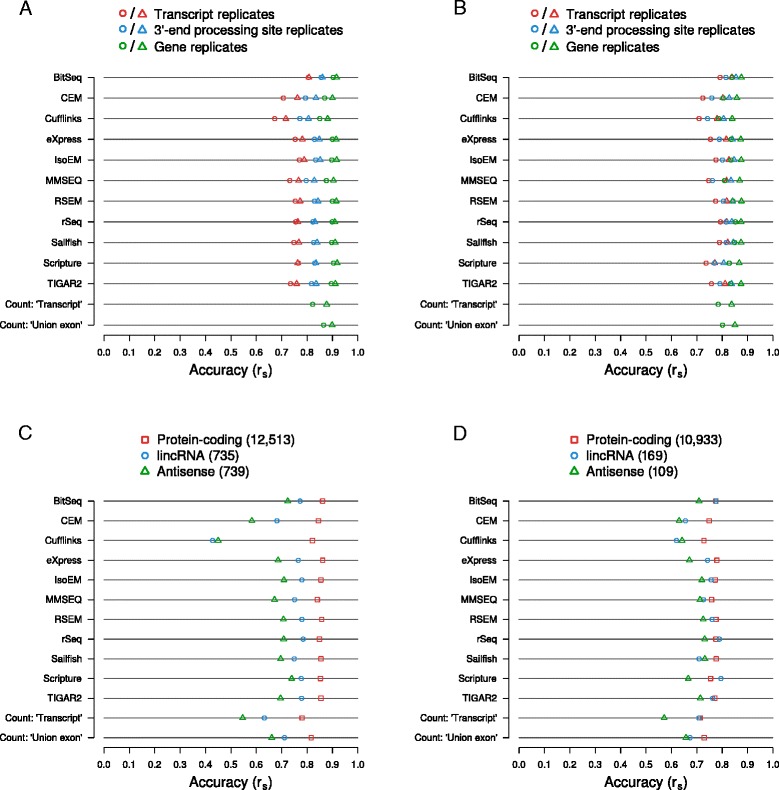
Results: We found that many tools have good accuracy and yield better estimates of gene-level expression compared to commonly used count-based approaches, but they vary widely in memory and runtime requirements. Nucleotide composition and intron/exon structure have comparatively little influence on the accuracy of expression estimates, which correlates most strongly with transcript/gene expression levels. To facilitate the reproduction and further extension of our study, we provide datasets, source code, and an online analysis tool on a companion website, where developers can upload expression estimates obtained with their own tool to compare them to those inferred by the methods assessed here.
Conclusions: As many methods for quantifying isoform abundance with comparable accuracy are available, a user's choice will likely be determined by factors such as the memory and runtime requirements, as well as the availability of methods for downstream analyses. Sequencing-based methods to quantify the abundance of specific transcript regions could complement validation schemes based on synthetic data and quantitative PCR in future or ongoing assessments of RNA-seq analysis methods.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
