

Whole exome sequencing in extended families with autism spectrum disorder implicates four candidate genes
- PMID: 26204995
- PMCID: PMC4578871
- DOI: 10.1007/s00439-015-1585-y
Whole exome sequencing in extended families with autism spectrum disorder implicates four candidate genes
Abstract
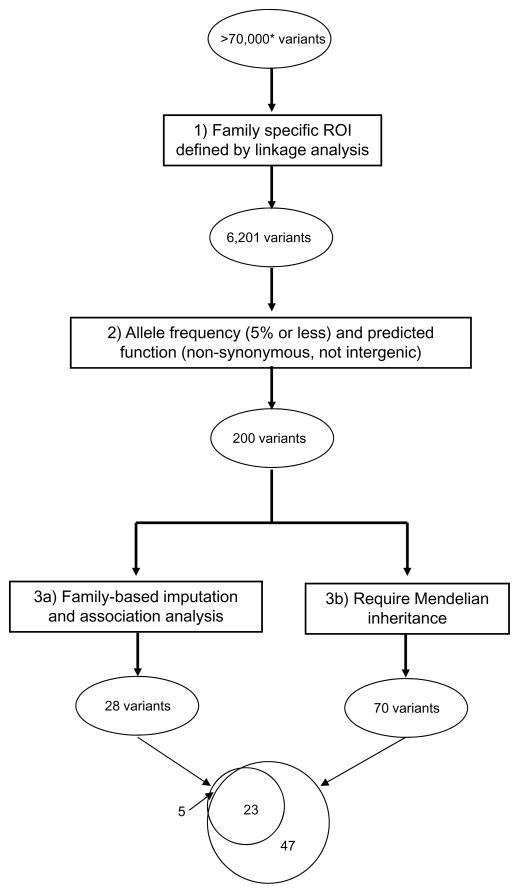
Autism spectrum disorders (ASDs) are a group of neurodevelopmental disorders, characterized by impairment in communication and social interactions, and by repetitive behaviors. ASDs are highly heritable, and estimates of the number of risk loci range from hundreds to >1000. We considered 7 extended families (size 12-47 individuals), each with ≥3 individuals affected by ASD. All individuals were genotyped with dense SNP panels. A small subset of each family was typed with whole exome sequence (WES). We used a 3-step approach for variant identification. First, we used family-specific parametric linkage analysis of the SNP data to identify regions of interest. Second, we filtered variants in these regions based on frequency and function, obtaining exactly 200 candidates. Third, we compared two approaches to narrowing this list further. We used information from the SNP data to impute exome variant dosages into those without WES. We regressed affected status on variant allele dosage, using pedigree-based kinship matrices to account for relationships. The p value for the test of the null hypothesis that variant allele dosage is unrelated to phenotype was used to indicate strength of evidence supporting the variant. A cutoff of p = 0.05 gave 28 variants. As an alternative third filter, we required Mendelian inheritance in those with WES, resulting in 70 variants. The imputation- and association-based approach was effective. We identified four strong candidate genes for ASD (SEZ6L, HISPPD1, FEZF1, SAMD11), all of which have been previously implicated in other studies, or have a strong biological argument for their relevance.
Figures
References
-
- American Psychiatric Association . Diagnostic and Statistical Manual. 5. American Psychiatric Association; Washington, DC: 2013.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
