

Molecular docking and structure-based drug design strategies
- PMID: 26205061
- PMCID: PMC6332083
- DOI: 10.3390/molecules200713384
Molecular docking and structure-based drug design strategies
Abstract
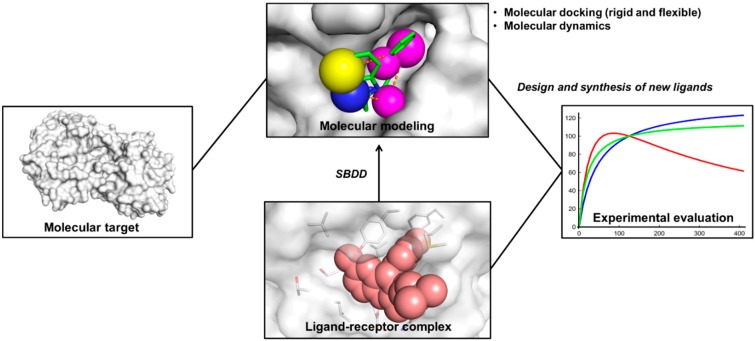
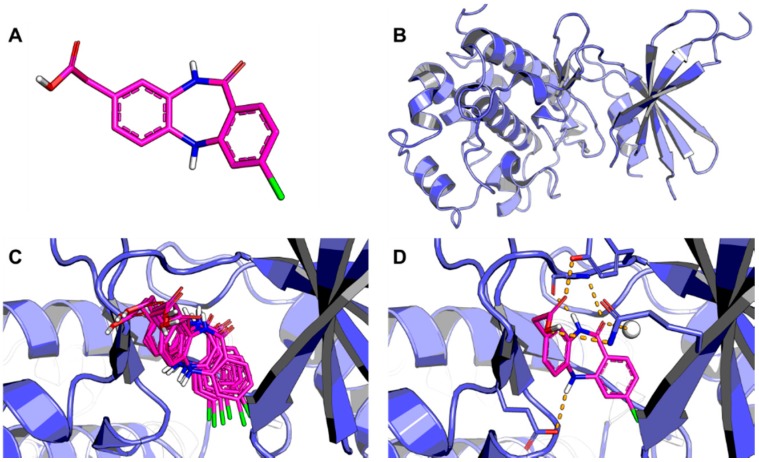
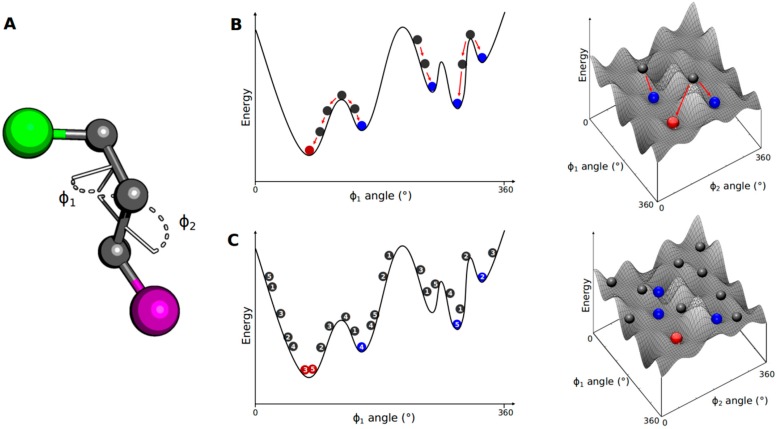
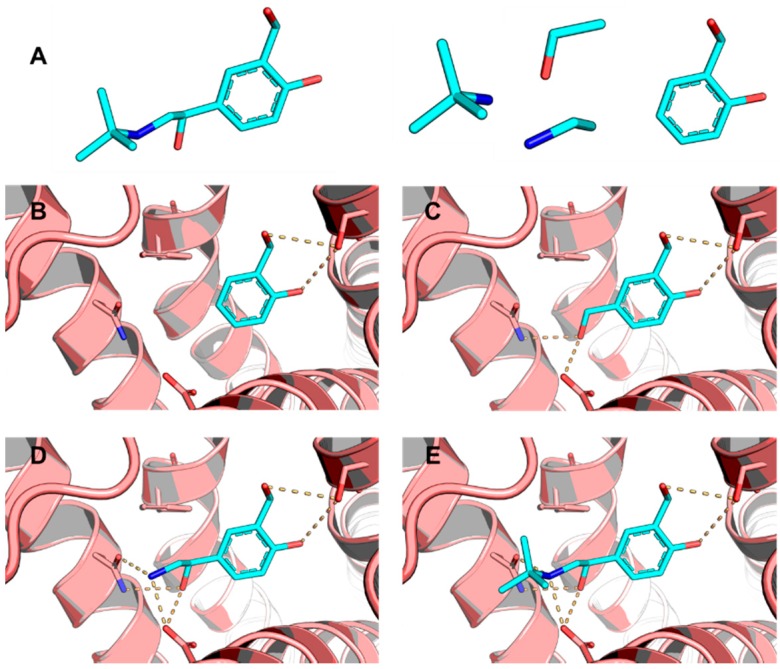
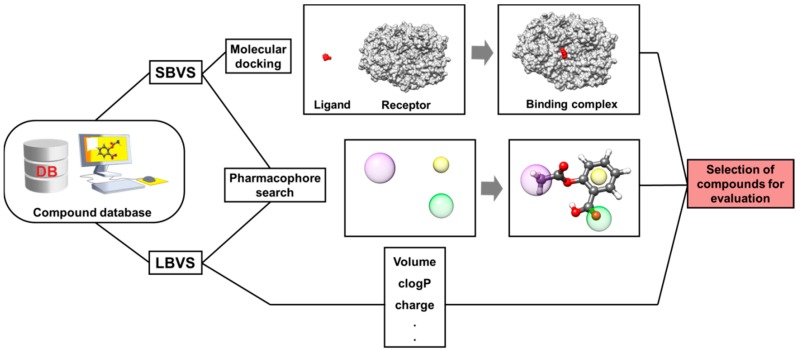
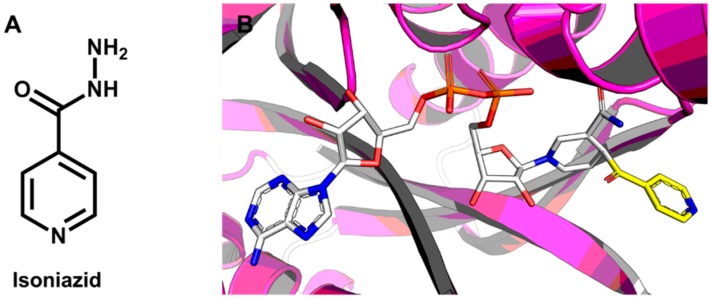
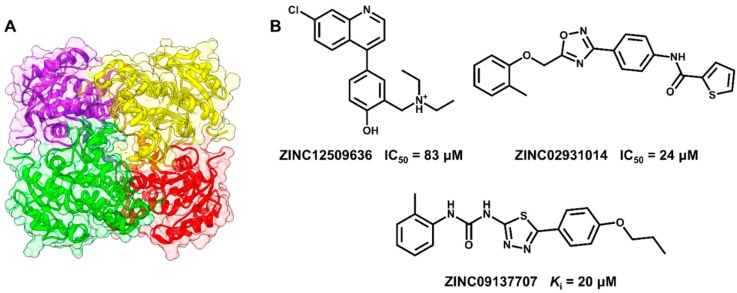
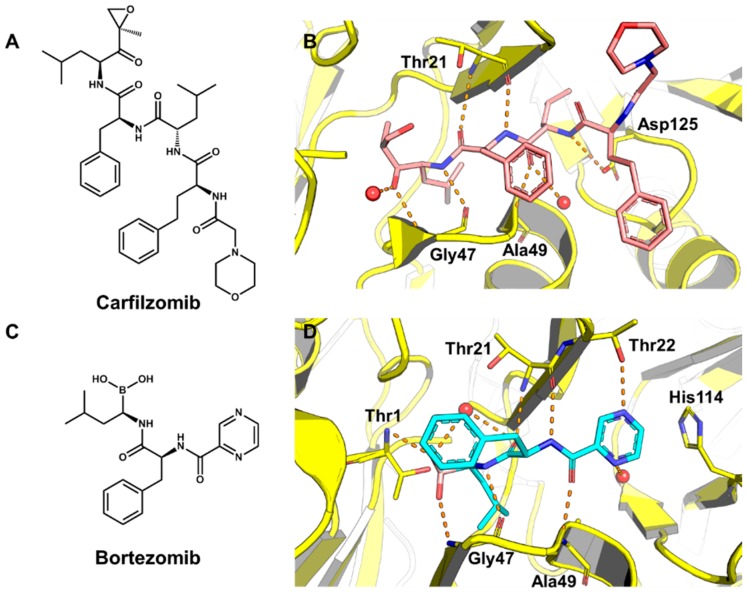
Pharmaceutical research has successfully incorporated a wealth of molecular modeling methods, within a variety of drug discovery programs, to study complex biological and chemical systems. The integration of computational and experimental strategies has been of great value in the identification and development of novel promising compounds. Broadly used in modern drug design, molecular docking methods explore the ligand conformations adopted within the binding sites of macromolecular targets. This approach also estimates the ligand-receptor binding free energy by evaluating critical phenomena involved in the intermolecular recognition process. Today, as a variety of docking algorithms are available, an understanding of the advantages and limitations of each method is of fundamental importance in the development of effective strategies and the generation of relevant results. The purpose of this review is to examine current molecular docking strategies used in drug discovery and medicinal chemistry, exploring the advances in the field and the role played by the integration of structure- and ligand-based methods.
Keywords: SBDD; SBVS; drug discovery; molecular interaction; molecular modeling; molecular target; pharmacophore; virtual screening.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
