

Complete loss of the DNAJB6 G/F domain and novel missense mutations cause distal-onset DNAJB6 myopathy
- PMID: 26205529
- PMCID: PMC4513909
- DOI: 10.1186/s40478-015-0224-0
Complete loss of the DNAJB6 G/F domain and novel missense mutations cause distal-onset DNAJB6 myopathy
Abstract
Introduction: Protein aggregation is a common cause of neuropathology. The protein aggregation myopathy Limb-Girdle Muscular Dystrophy 1D (LGMD1D) is caused by mutations of amino acids Phe89 or Phe93 of DNAJB6, a co-chaperone of the HSP70 anti-aggregation protein. Another DNAJB6 mutation, Pro96Arg, was found to cause a distal-onset myopathy in one family.
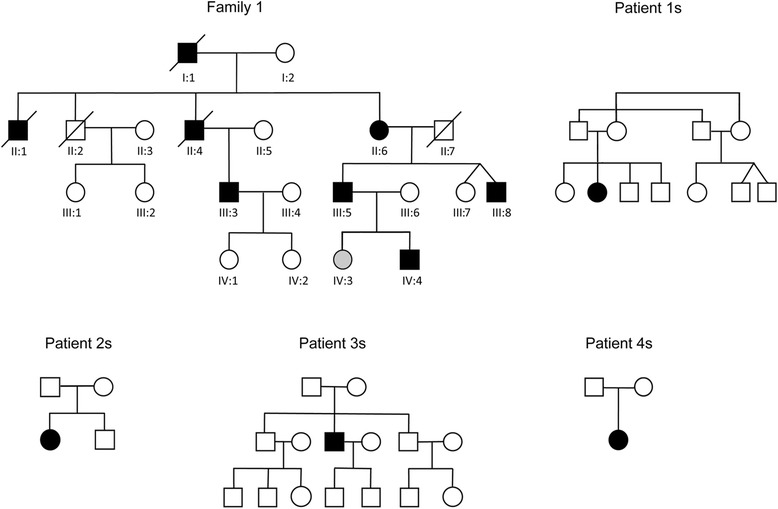
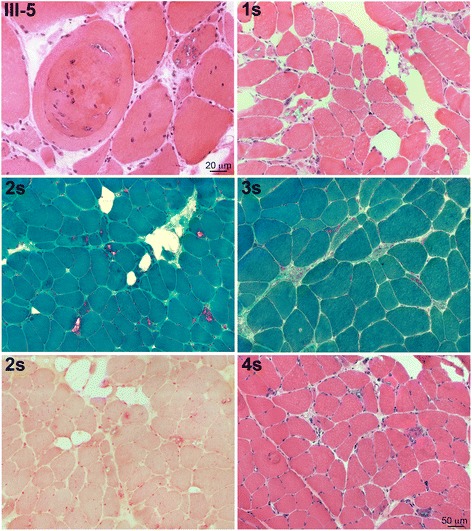
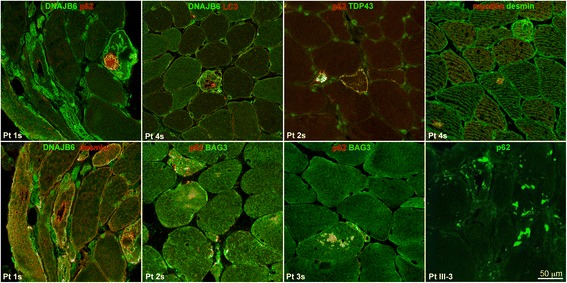
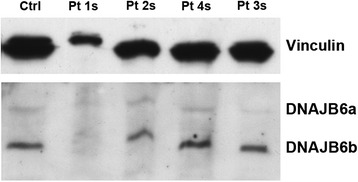
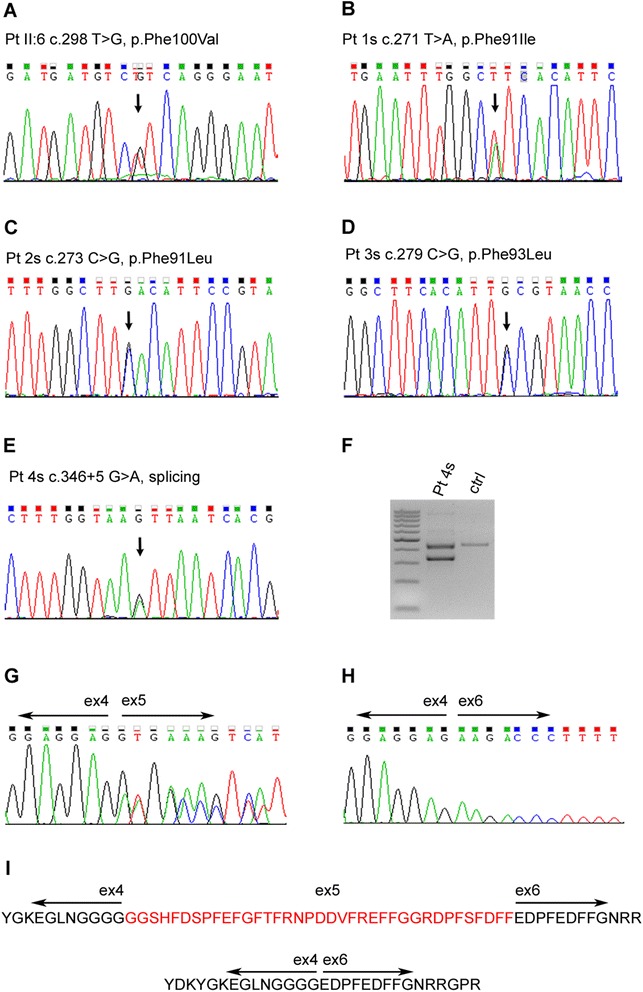
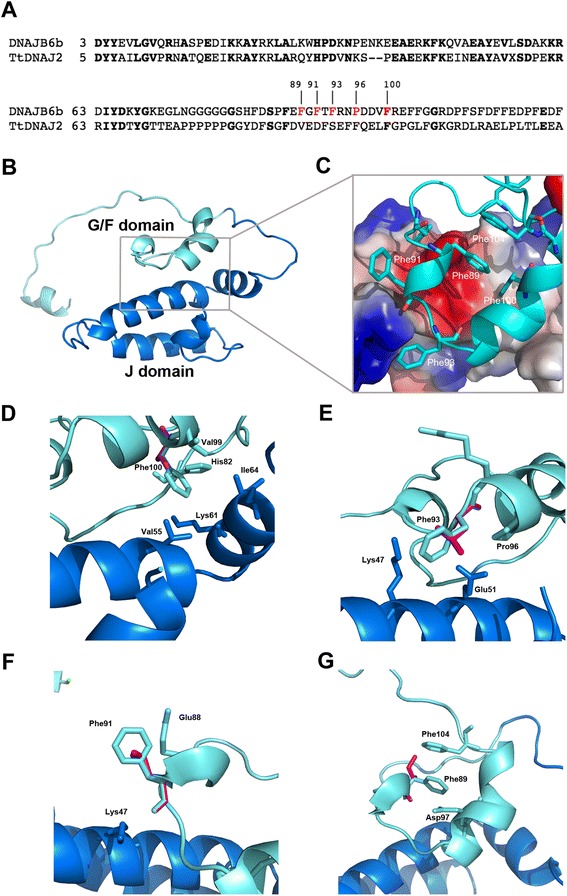
Results: We detail the mutational, neuropathological, neurophysiological, neurological and radiological features of five new DNAJB6-myopathy families. One has the known Phe93Leu mutation and classic late-onset slowly progressive LGMD1D. Two have different mutations of Phe91 causing a variant childhood-onset severe limb-girdle myopathy. One has a Phe100Val mutation and distal-onset myopathy, unique early bulbar involvement, and a gender-modified wide age-of-onset range. The last has childhood-onset severe distal-onset myopathy and the first non-missense DNAJB6 mutation, c.346 + 5G > A, causing a splicing defect that entirely eliminates DNAJB6's G/F domain (ΔG/F), the domain that harbours all other mutations. Clinical and imaging examinations reveal that muscles considered uninvolved in DNAJB6-myopathy, e.g. lateral gastrocnemii, are affected in our patients with new mutations. Mutational modelling based on the known structure of the bacterial DNAJ2 protein indicates that all past and present mutated residues cluster within 15 Å in the G/F domain and all disturb the interface of this domain with the protein's J domain that confers the interaction with HSP70.
Conclusions: Our patients expand the phenotypic spectrum of DNAJB6-myopathy and allow tentative genotype-phenotype specifications. Combining with previous studies, the clinical severity spectrum is as follows: ΔG/F and Phe91 mutations, most severe; Phe100, Pro96, Phe89 mutations, intermediate; and Phe93, least severe. As it stands presently, proximal G/F domain mutations (Phe89, Phe91, Phe93) cause proximal limb-girdle myopathy, while distal G/F mutations (Pro96, Phe100) cause distal-onset myopathy. While all mutations affect the G/F-J interaction, each likely does so in different unknown extents or ways. One mutation, ΔG/F, causes its associated severe distal-onset myopathy phenotype in a clear way, through generation of a G/F domain-lacking DNAJB6 protein.
Figures
References
-
- Sarparanta J, Jonson PH, Golzio C, Sandell S, Luque H, Screen M, McDonald K, Stajich JM, Mahjneh I, Vihola A, Hatakeyama S, Tanaka S, Sasaki H. Raheem O, Penttilä S, Lehtinen S, Huovinen S, Palmio J, Tasca G, Ricci E, Hackman P, Hauser M, Katsanis N, Udd B (2012) Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet 44(450–455):S451–S452. doi:10.1038/ng.1103 - PMC - PubMed
-
- Couthouis J, Raphael AR, Siskind C, Findlay AR, Buenrostro JD, Greenleaf WJ, Vogel H, Day JW, Flanigan KM, Gitler AD. Exome sequencing identifies a DNAJB6 mutation in a family with dominantly-inherited limb-girdle muscular dystrophy. Neuromuscul Disord. 2014;24:431–435. doi: 10.1016/j.nmd.2014.01.014. - DOI - PMC - PubMed
-
- Sato T, Hayashi YK, Oya Y, Kondo T, Sugie K, Kaneda D, Houzen H, Yabe I, Sasaki H, Noguchi S, Nonaka I, Osawa M, Nishino I (2013) DNAJB6 myopathy in an Asian cohort and cytoplasmic/nuclear inclusions. Neuromuscul Disord 23:269–276. doi:10.1016/j.nmd.2012.12.010 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
