

Mutations in Plasmalemma Vesicle Associated Protein Result in Sieving Protein-Losing Enteropathy Characterized by Hypoproteinemia, Hypoalbuminemia, and Hypertriglyceridemia
- PMID: 26207260
- PMCID: PMC4507283
- DOI: 10.1016/j.jcmgh.2015.05.001
Mutations in Plasmalemma Vesicle Associated Protein Result in Sieving Protein-Losing Enteropathy Characterized by Hypoproteinemia, Hypoalbuminemia, and Hypertriglyceridemia
Abstract
Background & aims methods: Severe intestinal diseases observed in very young children are often the result of monogenic defects. We used whole exome sequencing (WES) to examine the genetic cause in a patient with a distinct severe form of protein losing enteropathy (PLE) characterized by hypoproteinemia, hypoalbuminemia, and hypertriglyceridemia.
Methods: WES was performed at the Centre for Applied Genomics, Hospital for Sick Children, Toronto, Canada. Exome library preparation was performed using the Ion Torrent AmpliSeq RDY Exome Kit. Functional studies were carried out based on the identified mutation.
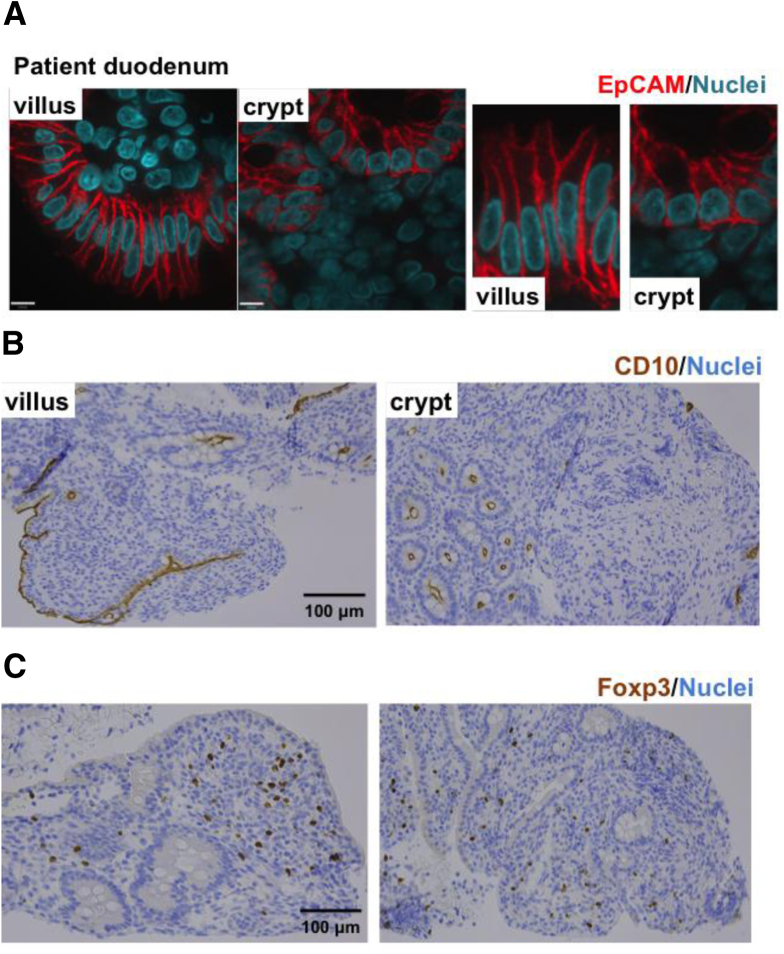
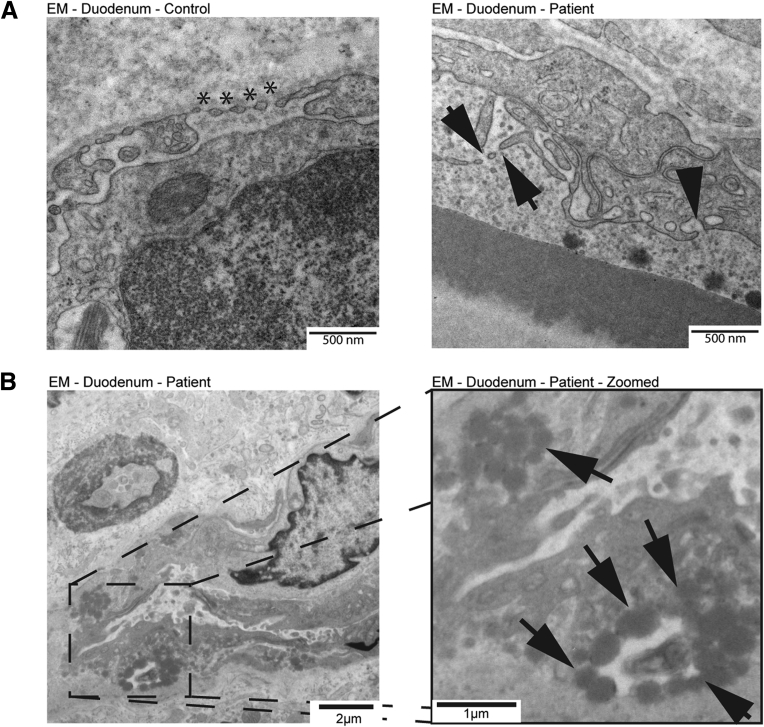
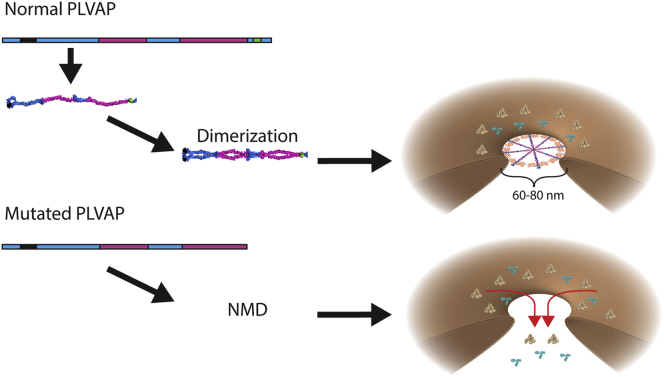
Results: Using whole exome sequencing we identified a homozygous nonsense mutation (1072C>T; p.Arg358*) in the PLVAP (plasmalemma vesicle associated protein) gene in an infant from consanguineous parents who died at five months of age of severe protein losing enteropathy. Functional studies determined that the mutated PLVAP mRNA and protein were not expressed in the patient biopsy tissues, presumably secondary to nonsense-mediated mRNA decay. Pathological analysis showed that the loss of PLVAP resulted in disruption of endothelial fenestrated diaphragms.
Conclusions: PLVAP p.Arg358* mutation resulted in loss of PLVAP expression with subsequent deletion of the diaphragms of endothelial fenestrae leading to plasma protein extravasation, protein-losing enteropathy and ultimately death.
Keywords: IBD; PLE; PLVAP; Protein losing enteropathy; VEOIBD; and hypertriglyceridemia; endothelium; fenestrae; hypoalbuminemia; hypoproteinemia; monogenic diseases; very early onset IBD.
Conflict of interest statement
The authors have no potential conflicts including financial, professional, or personal that are relevant to the manuscript.
Figures









References
-
- Umar S.B., DiBaise J.K. Protein-losing enteropathy: case illustrations and clinical review. Am J Gastroenterol. 2010;105:43–49. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases