

Expanding the Spectrum of Founder Mutations Causing Isolated Gonadotropin-Releasing Hormone Deficiency
- PMID: 26207952
- PMCID: PMC4596034
- DOI: 10.1210/jc.2015-2262
Expanding the Spectrum of Founder Mutations Causing Isolated Gonadotropin-Releasing Hormone Deficiency
Abstract
Context: Loss of function (LoF) mutations in more than 20 genes are now known to cause isolated GnRH deficiency (IGD) in humans. Most causal IGD mutations are typically private, ie, limited to a single individual/pedigree. However, somewhat paradoxically, four IGD genes (GNRH1, TAC3, PROKR2, and GNRHR) have been shown to harbor LoF founder mutations that are shared by multiple unrelated individuals. It is not known whether similar founder mutations occur in other IGD genes.
Objective: The objective of the study was to determine whether shared deleterious mutations in IGD-associated genes represent founder alleles.
Setting: This study was an international collaboration among academic medical centers.
Methods: IGD patients with shared mutations, defined as those documented in three or more unrelated probands in 14 IGD-associated genes, were identified from various academic institutions, the Human Gene Mutation Database, and literature reports by other international investigators. Haplotypes of single-nucleotide polymorphisms and short tandem repeats surrounding the mutations were constructed to assess genetic ancestry.
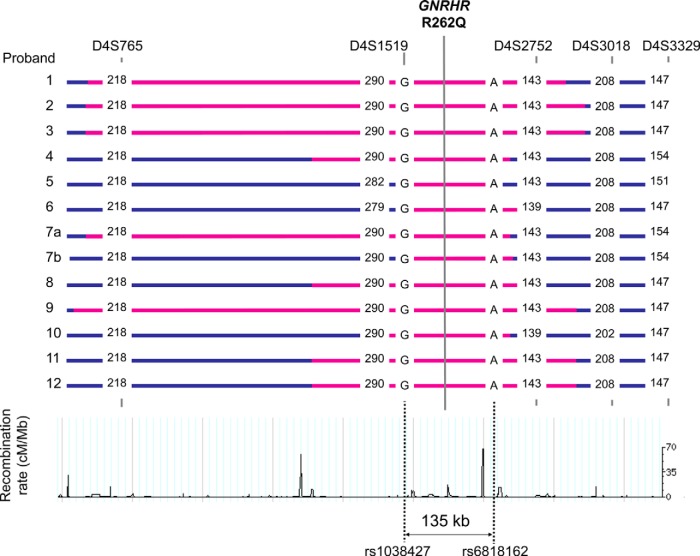
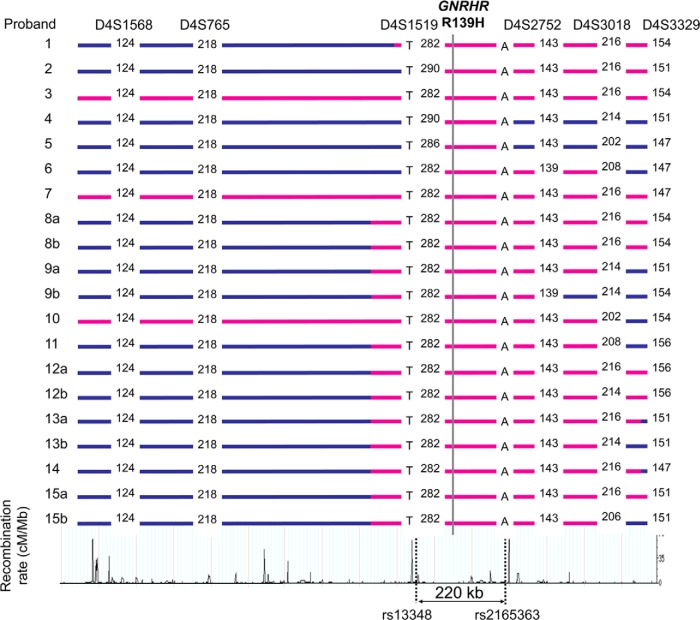
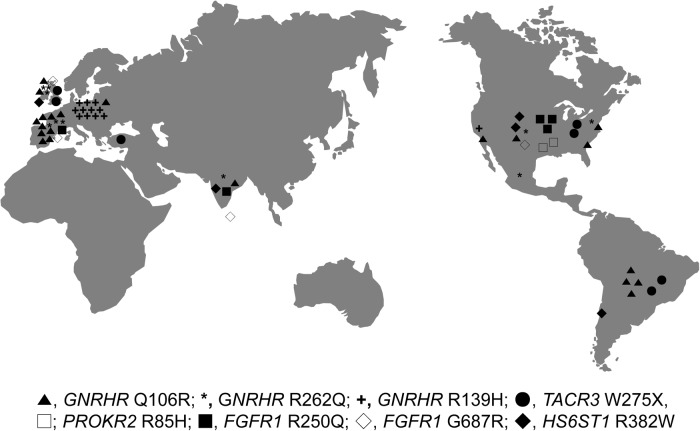
Results: A total of eight founder mutations in five genes, GNRHR (Q106R, R262Q, R139H), TACR3 (W275X), PROKR2 (R85H), FGFR1 (R250Q, G687R), and HS6ST1 (R382W) were identified. Most founder alleles were present at low frequency in the general population. The estimated age of these mutant alleles ranged from 1925 to 5600 years and corresponded to the time of rapid human population expansion.
Conclusions: We have expanded the spectrum of founder alleles associated with IGD to a total of eight founder mutations. In contrast to the approximately 9000-year-old PROKR2 founder allele that may confer a heterozygote advantage, the rest of the founder alleles are relatively more recent in origin, in keeping with the timing of recent human population expansion and any selective heterozygote advantage of these alleles requires further evaluation.
Figures
Similar articles
-
A genetic basis for functional hypothalamic amenorrhea.N Engl J Med. 2011 Jan 20;364(3):215-25. doi: 10.1056/NEJMoa0911064. N Engl J Med. 2011. PMID: 21247312 Free PMC article.
-
An ancient founder mutation in PROKR2 impairs human reproduction.Hum Mol Genet. 2012 Oct 1;21(19):4314-24. doi: 10.1093/hmg/dds264. Epub 2012 Jul 5. Hum Mol Genet. 2012. PMID: 22773735 Free PMC article.
-
Modeling mutant/wild-type interactions to ascertain pathogenicity of PROKR2 missense variants in patients with isolated GnRH deficiency.Hum Mol Genet. 2018 Jan 15;27(2):338-350. doi: 10.1093/hmg/ddx404. Hum Mol Genet. 2018. PMID: 29161432 Free PMC article.
-
Non-syndromic congenital hypogonadotropic hypogonadism: clinical presentation and genotype-phenotype relationships.Eur J Endocrinol. 2010 May;162(5):835-51. doi: 10.1530/EJE-10-0083. Epub 2010 Mar 5. Eur J Endocrinol. 2010. PMID: 20207726 Review.
-
GnRH receptor mutations in isolated gonadotropic deficiency.Mol Cell Endocrinol. 2011 Oct 22;346(1-2):21-8. doi: 10.1016/j.mce.2011.04.018. Epub 2011 Apr 30. Mol Cell Endocrinol. 2011. PMID: 21645587 Review.
Cited by
-
Functional Hypogonadotropic Hypogonadism in Men: Underlying Neuroendocrine Mechanisms and Natural History.J Clin Endocrinol Metab. 2019 Aug 1;104(8):3403-3414. doi: 10.1210/jc.2018-02697. J Clin Endocrinol Metab. 2019. PMID: 31220265 Free PMC article.
-
Genetic evaluation supports differential diagnosis in adolescent patients with delayed puberty.Eur J Endocrinol. 2021 Oct 8;185(5):617-627. doi: 10.1530/EJE-21-0387. Eur J Endocrinol. 2021. PMID: 34403359 Free PMC article.
-
From Steroid and Drug Metabolism to Glycobiology, Using Sulfotransferase Structures to Understand and Tailor Function.Drug Metab Dispos. 2022 Jul;50(7):1027-1041. doi: 10.1124/dmd.121.000478. Epub 2022 Feb 22. Drug Metab Dispos. 2022. PMID: 35197313 Free PMC article. Review.
-
High Population Frequency of GNRHR p.Q106R in Malta: An Evaluation of Fertility and Hormone Profiles in Heterozygotes.J Endocr Soc. 2023 Dec 29;8(2):bvad172. doi: 10.1210/jendso/bvad172. eCollection 2024 Jan 5. J Endocr Soc. 2023. PMID: 38196663 Free PMC article.
-
Mutations in gonadotropin-releasing hormone signaling pathway in two nIHH patients with successful pregnancy outcomes.Reprod Biol Endocrinol. 2016 Aug 20;14(1):48. doi: 10.1186/s12958-016-0183-8. Reprod Biol Endocrinol. 2016. PMID: 27544332 Free PMC article.
References
-
- Bouligand J, Ghervan C, Tello JA, et al. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 2009;360:2742–2748. - PubMed
-
- Young J, Bouligand J, Francou B, et al. TAC3 and TACR3 defects cause hypothalamic congenital hypogonadotropic hypogonadism in humans. J Clin Endocrinol Metab. 2010;95:2287–2295. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
