

Cross-cancer profiling of molecular alterations within the human autophagy interaction network
- PMID: 26208877
- PMCID: PMC4590660
- DOI: 10.1080/15548627.2015.1067362
Cross-cancer profiling of molecular alterations within the human autophagy interaction network
Abstract
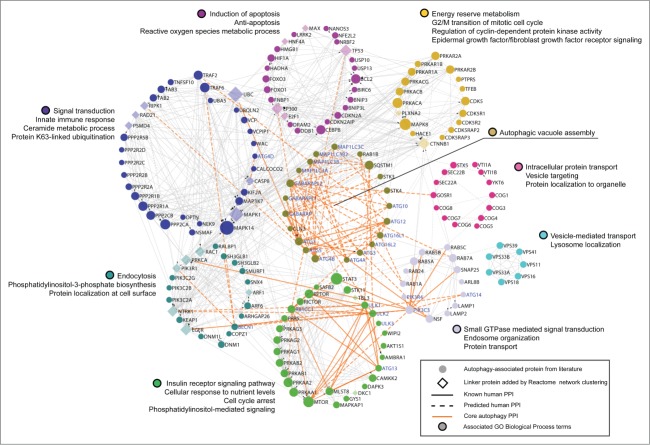
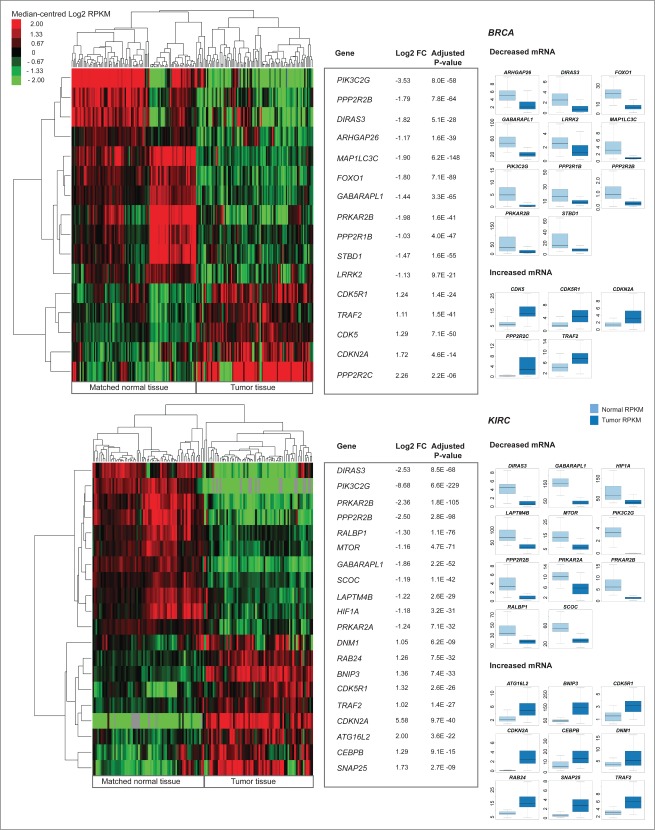
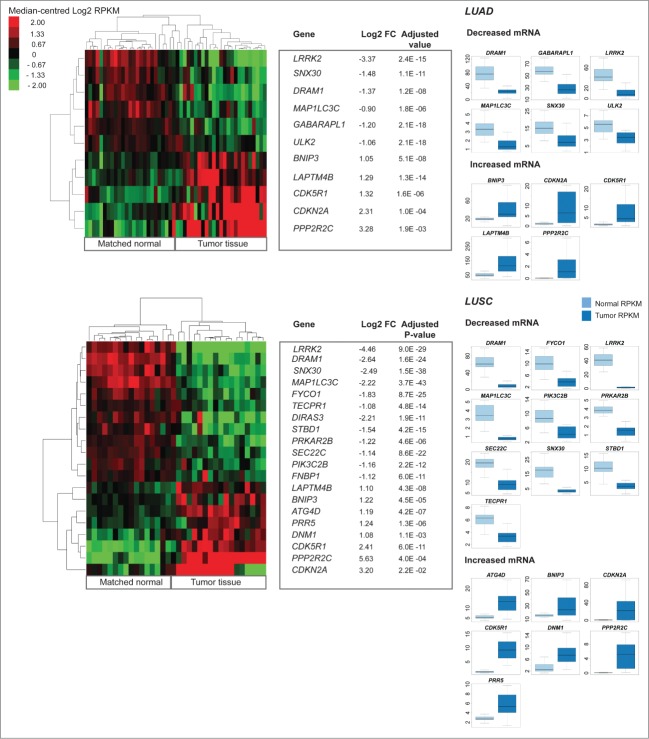
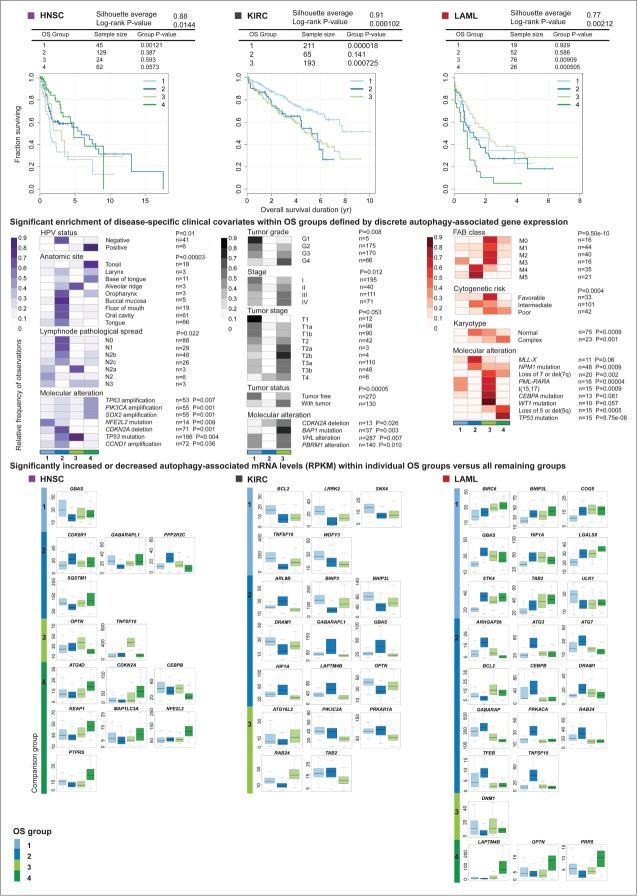
Aberrant activation or disruption of autophagy promotes tumorigenesis in various preclinical models of cancer, but whether the autophagy pathway is a target for recurrent molecular alteration in human cancer patient samples is unknown. To address this outstanding question, we surveyed 211 human autophagy-associated genes for tumor-related alterations to DNA sequence and RNA expression levels and examined their association with patient survival outcomes in multiple cancer types with sequence data from The Cancer Genome Atlas consortium. We found 3 (RB1CC1/FIP200, ULK4, WDR45/WIPI4) and one (ATG7) core autophagy genes to be under positive selection for somatic mutations in endometrial carcinoma and clear cell renal carcinoma, respectively, while 29 autophagy regulators and pathway interactors, including previously identified KEAP1, NFE2L2, and MTOR, were significantly mutated in 6 of the 11 cancer types examined. Gene expression analyses revealed that GABARAPL1 and MAP1LC3C/LC3C transcripts were less abundant in breast cancer and non-small cell lung cancers than in matched normal tissue controls; ATG4D transcripts were increased in lung squamous cell carcinoma, as were ATG16L2 transcripts in kidney cancer. Unsupervised clustering of autophagy-associated mRNA levels in tumors stratified patient overall survival in 3 of 9 cancer types (acute myeloid leukemia, clear cell renal carcinoma, and head and neck cancer). These analyses provide the first comprehensive resource of recurrently altered autophagy-associated genes in human tumors, and highlight cancer types and subtypes where perturbed autophagy may be relevant to patient overall survival.
Keywords: AML; LAPTM4B; SNV; SQSTM1; TCGA; autophagy; cancer; gene expression; mutation; tumor.
Figures


References
-
- Leone RD, Amaravadi RK. Autophagy: a targetable linchpin of cancer cell metabolism. Trends Endocrinol Metab 2013; 24:209-17; PMID:23474062; http://dx.doi.org/10.1016/j.tem.2013.01.008 - DOI - PMC - PubMed
-
- Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10:1533-41; PMID:21878654; http://dx.doi.org/10.1158/1535-7163.MCT-11-0047 - DOI - PMC - PubMed
-
- Lebovitz CB, Bortnik SB, Gorski SM. Here, there be dragons: charting autophagy-related alterations in human tumors. Clin Cancer Res Off J Am Assoc Cancer Res 2012; 18:1214-26; PMID:22253413; http://dx.doi.org/10.1158/1078-0432.CCR-11-2465 - DOI - PubMed
-
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science 2013; 339:1546-58; PMID:23539594; http://dx.doi.org/10.1126/science.1235122 - DOI - PMC - PubMed
-
- Rung J, Brazma A. Reuse of public genome-wide gene expression data. Nat Rev Genet 2013; 14:89-99; PMID:23269463; http://dx.doi.org/10.1038/nrg3394 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous