

PoMo: An Allele Frequency-Based Approach for Species Tree Estimation
- PMID: 26209413
- PMCID: PMC4604832
- DOI: 10.1093/sysbio/syv048
PoMo: An Allele Frequency-Based Approach for Species Tree Estimation
Abstract
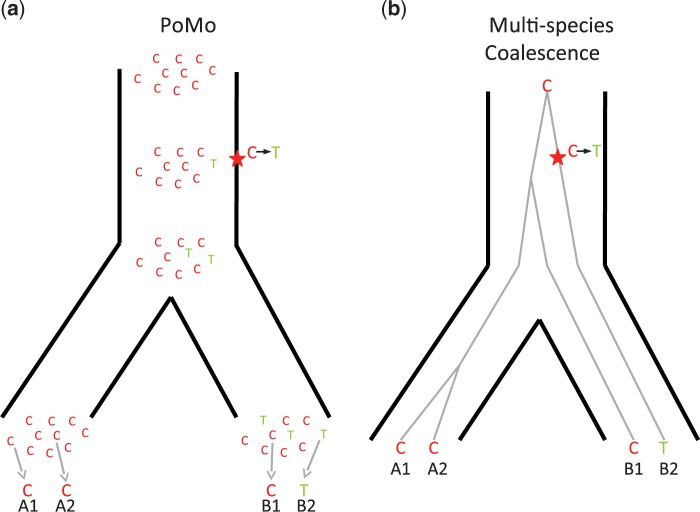
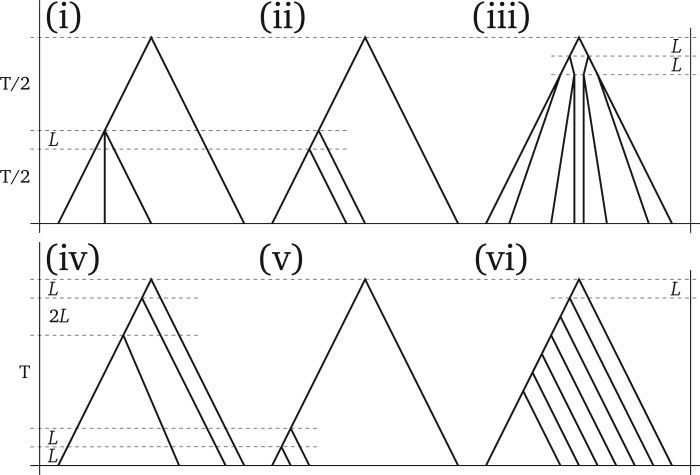
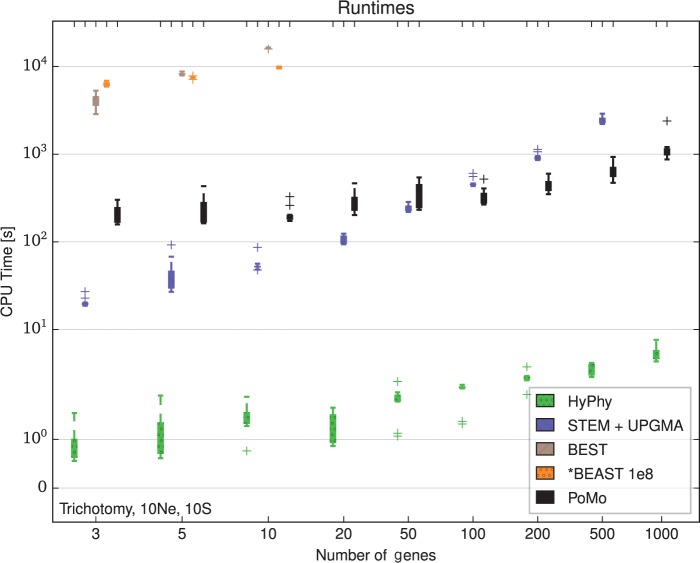
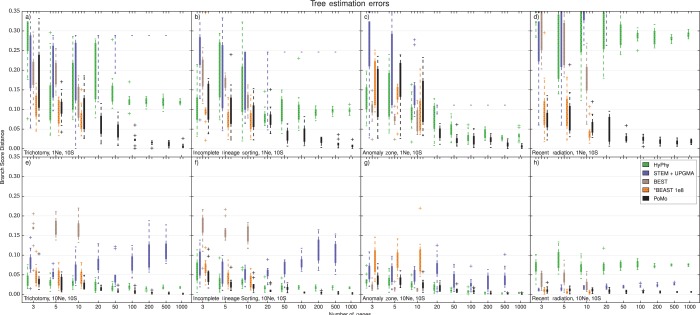
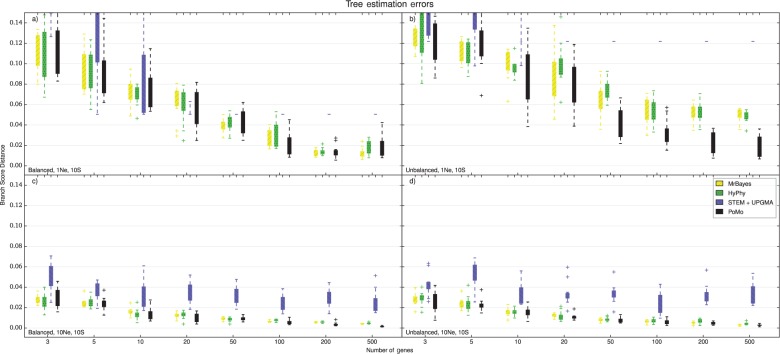
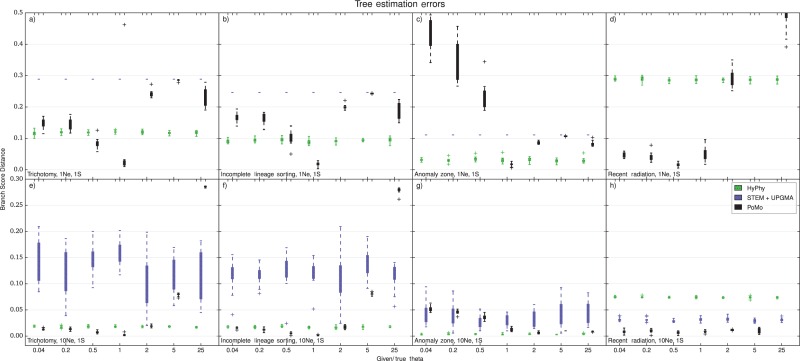
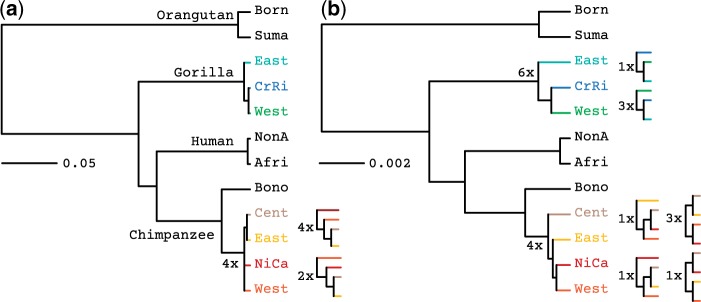
Incomplete lineage sorting can cause incongruencies of the overall species-level phylogenetic tree with the phylogenetic trees for individual genes or genomic segments. If these incongruencies are not accounted for, it is possible to incur several biases in species tree estimation. Here, we present a simple maximum likelihood approach that accounts for ancestral variation and incomplete lineage sorting. We use a POlymorphisms-aware phylogenetic MOdel (PoMo) that we have recently shown to efficiently estimate mutation rates and fixation biases from within and between-species variation data. We extend this model to perform efficient estimation of species trees. We test the performance of PoMo in several different scenarios of incomplete lineage sorting using simulations and compare it with existing methods both in accuracy and computational speed. In contrast to other approaches, our model does not use coalescent theory but is allele frequency based. We show that PoMo is well suited for genome-wide species tree estimation and that on such data it is more accurate than previous approaches.
Keywords: Incomplete lineage sorting; Phylogenetics; PoMo; Species tree.
© The Author(s) 2015. Published by Oxford University Press, on behalf of the Society of Systematic Biologists.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
