

Live imaging of endogenous Ca²⁺/calmodulin-dependent protein kinase II in neurons reveals that ischemia-related aggregation does not require kinase activity
- PMID: 26212614
- PMCID: PMC4636925
- DOI: 10.1111/jnc.13263
Live imaging of endogenous Ca²⁺/calmodulin-dependent protein kinase II in neurons reveals that ischemia-related aggregation does not require kinase activity
Abstract
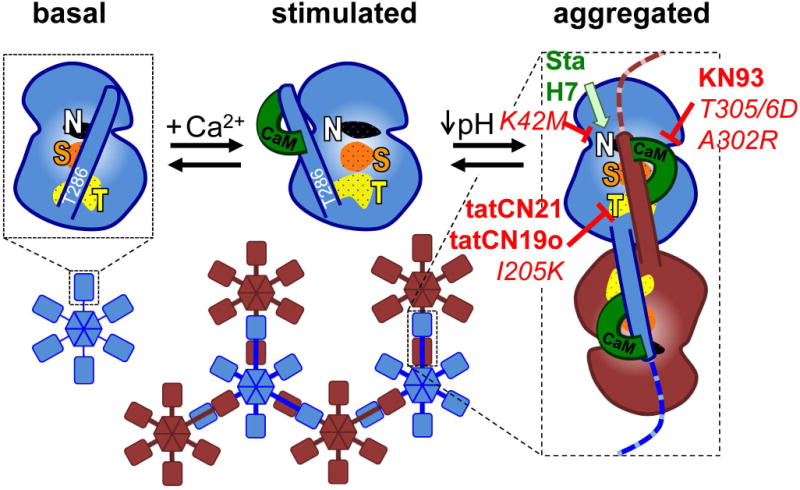
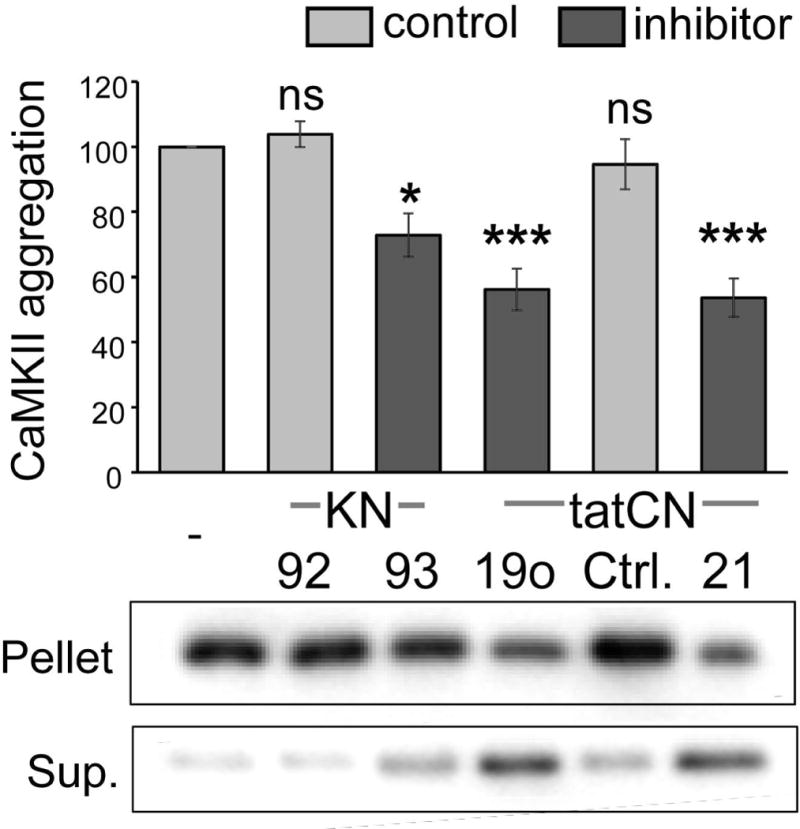
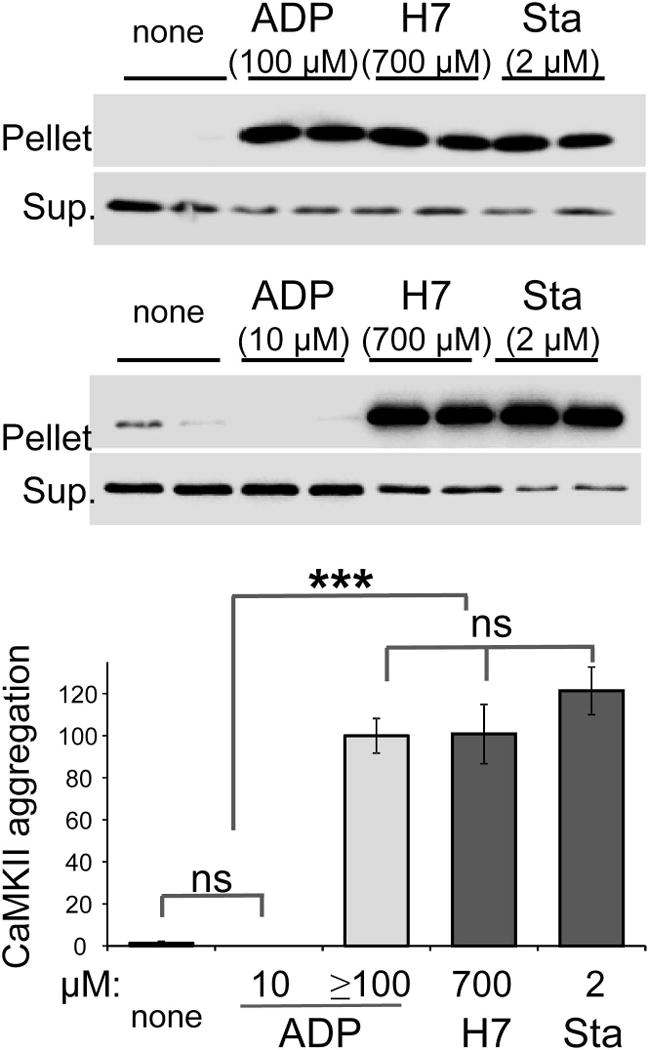
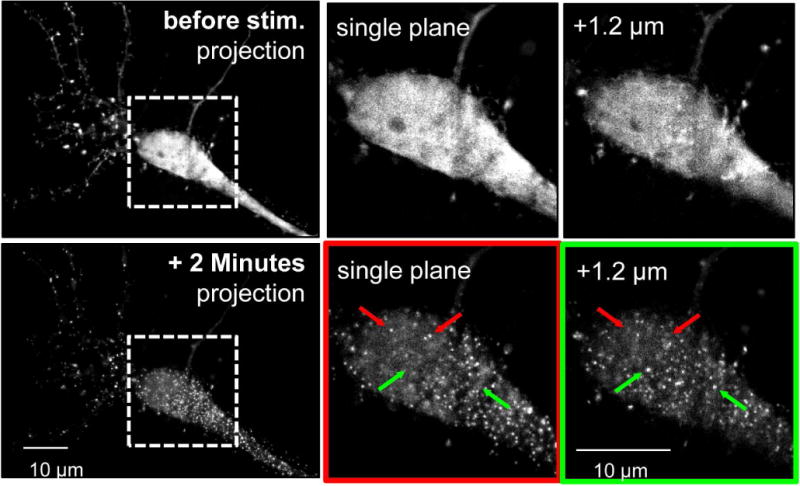
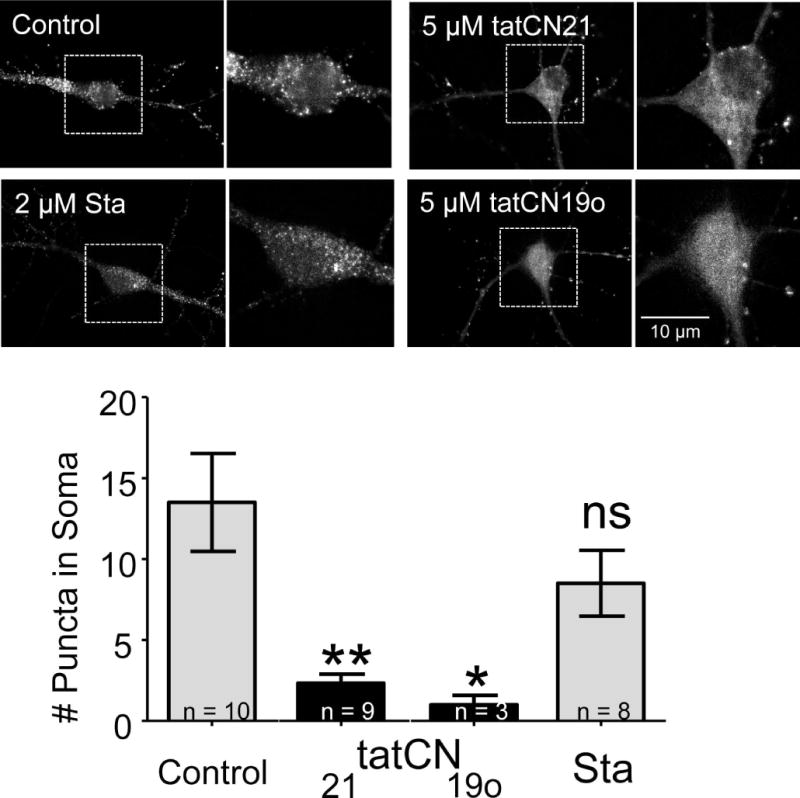
The Ca(2+) /calmodulin-dependent protein kinase II (CaMKII) forms 12meric holoenzymes. These holoenzymes cluster into larger aggregates within neurons under ischemic conditions and in vitro when ischemic conditions are mimicked. This aggregation is thought to be mediated by interaction between the regulatory domain of one kinase subunit with the T-site of another kinase subunit in a different holoenzyme, an interaction that requires stimulation by Ca(2+) /CaM and nucleotide for its induction. This model makes several predictions that were verified here: Aggregation in vitro was reduced by the CaMKII inhibitors tatCN21 and tatCN19o (which block the T-site) as well as by KN93 (which is CaM-competitive). Notably, these and previously tested manipulations that block CaMKII activation all reduced aggregation, suggesting an alternative mechanism that instead requires kinase activity. However, experiments with the nucleotide-competitive broad-spectrum kinase inhibitors staurosporin and H7 showed that this is not the case. In vitro, staurosporine and H7 enabled CaMKII aggregation even in the absence of nucleotide. Within rat hippocampal neurons, an intra-body enabled live monitoring of endogenous CaMKII aggregation. This aggregation was blocked by tatCN21, but not by staurosporine, even though both effectively inhibit CaMKII activity. These results support the mechanistic model for CaMKII aggregation and show that kinase activity is not required. CaMKII aggregation is prevented by inhibiting kinase activity with mutations (red italics; shown previously) or inhibitors (red bold; shown here), indicating requirement of kinase activity. However, we show here that nucleotide-competitive inhibitors (green) allow CaMKII aggregation (including endogenous CaMKII within neurons), demonstrating that kinase activity is not required and supporting the current mechanistic model for CaMKII aggregation.
Keywords: CaMKII; FingR; aggregation; glutamate; hippocampus; ischemia.
© 2015 International Society for Neurochemistry.
Conflict of interest statement
Figures
References
-
- Aarts MM, Tymianski M. Molecular mechanisms underlying specificity of excitotoxic signaling in neurons. Curr Mol Med. 2004;4:137–147. - PubMed
-
- Ashpole NM, Hudmon A. Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol Cell Neurosci. 2011;46:720–730. - PubMed
-
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- F31NS083298/NS/NINDS NIH HHS/United States
- R01NS081248/NS/NINDS NIH HHS/United States
- T32 NS099042/NS/NINDS NIH HHS/United States
- T32 HD041697/HD/NICHD NIH HHS/United States
- R01 NS081248/NS/NINDS NIH HHS/United States
- R01NS080851/NS/NINDS NIH HHS/United States
- P30 NS048154/NS/NINDS NIH HHS/United States
- R01 NS080851/NS/NINDS NIH HHS/United States
- F31 NS083298/NS/NINDS NIH HHS/United States
- F31 NS092265/NS/NINDS NIH HHS/United States
- T32HD041697/HD/NICHD NIH HHS/United States
- F31NS092265/NS/NINDS NIH HHS/United States
- P30NS048154/NS/NINDS NIH HHS/United States
- R01 NS081678/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
