

Mitochondrial CB1 receptor is involved in ACEA-induced protective effects on neurons and mitochondrial functions
- PMID: 26215450
- PMCID: PMC4516969
- DOI: 10.1038/srep12440
Mitochondrial CB1 receptor is involved in ACEA-induced protective effects on neurons and mitochondrial functions
Abstract
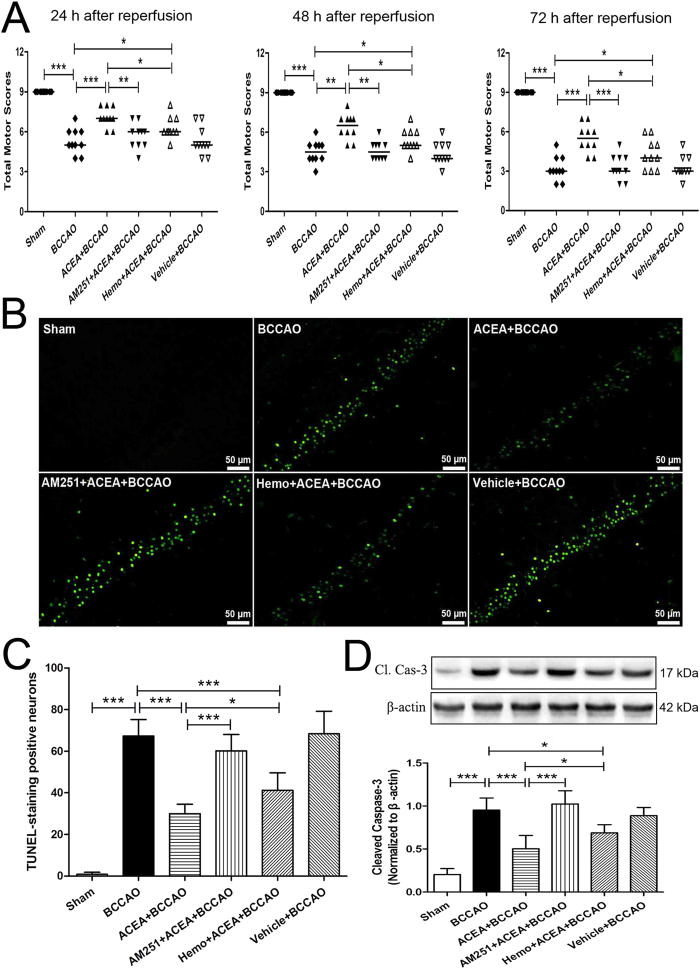
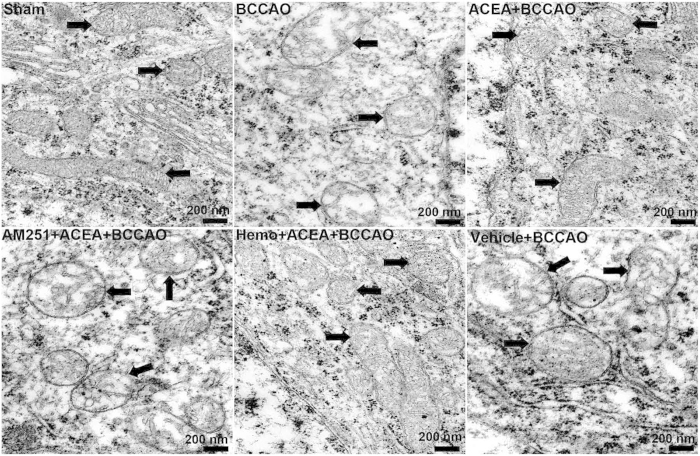
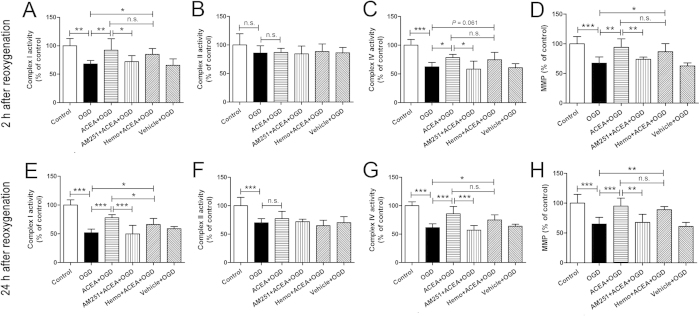
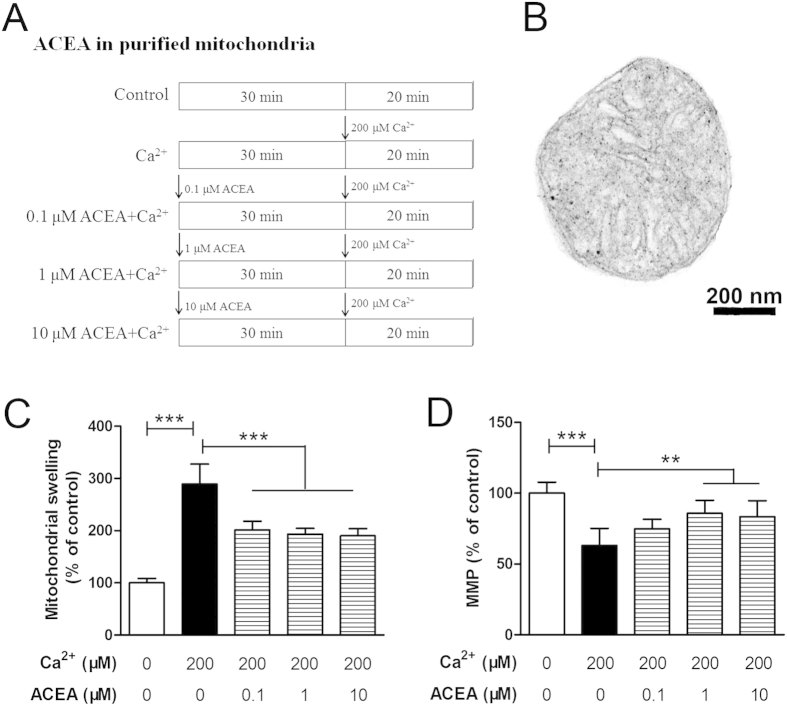
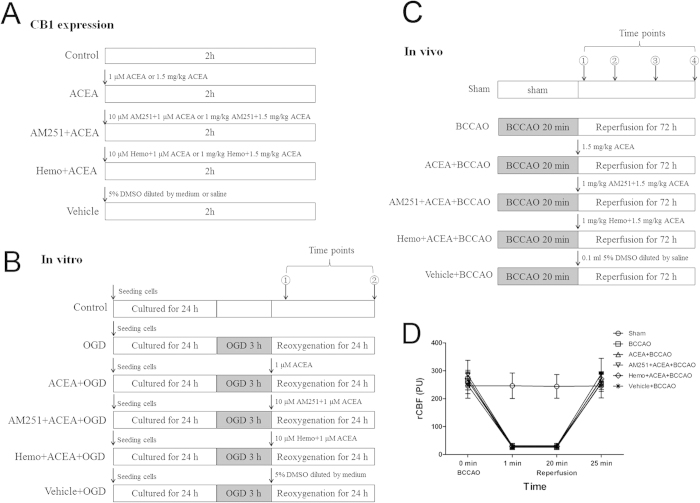
Mitochondrial dysfunction contributes to cell death after cerebral ischemia/reperfusion (I/R) injury. Cannabinoid CB1 receptor is expressed in neuronal mitochondrial membranes (mtCB1R) and involved in regulating mitochondrial functions under physiological conditions. However, whether mtCB1R affords neuroprotection against I/R injury remains unknown. We used mouse models of cerebral I/R, primary cultured hippocampal neurons exposed to oxygen-glucose deprivation/reoxygenation (OGD/R) and Ca(2+)-induced injury in purified neuronal mitochondria to investigate the role of mtCB1R in neuroprotection. Our results showed selective cell-permeant CB1 receptor agonist, arachidonyl-2-chloroethylamide (ACEA), significantly up-regulated the expression of mtCB1R protein in hippocampal neurons and tissue. In vitro, ACEA restored cell viability, inhibited generation of reactive oxygen species (ROS), decreased lactate dehydrogenase (LDH) release and reduced apoptosis, improved mitochondrial function. In vivo, ACEA ameliorated neurological scores, diminished the number of TUNEL-positive neurons and decreased the expression of cleaved caspase-3. However, ACEA-induced benefits were blocked by the selective cell-permeant CB1 receptor antagonist AM251, but just partially by the selective cell-impermeant CB1 receptor antagonist hemopressin. In purified neuronal mitochondria, mtCB1R activation attenuated Ca(2+)-induced mitochondrial injury. In conclusion, mtCB1R is involved in ACEA-induced protective effects on neurons and mitochondrial functions, suggesting mtCB1R may be a potential novel target for the treatment of brain ischemic injury.
Figures
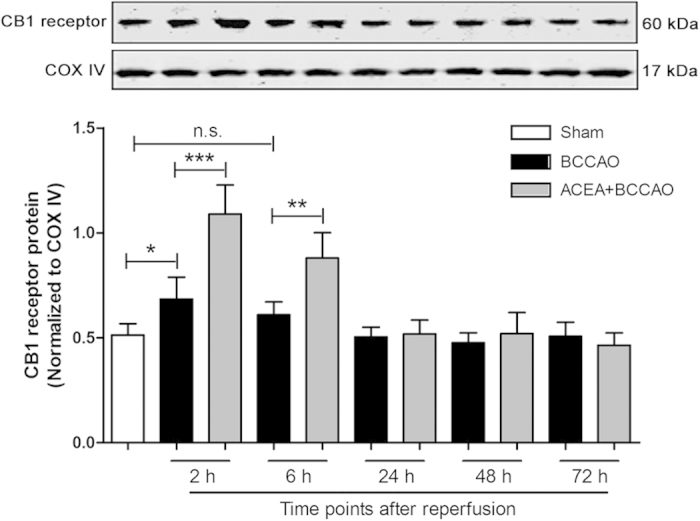
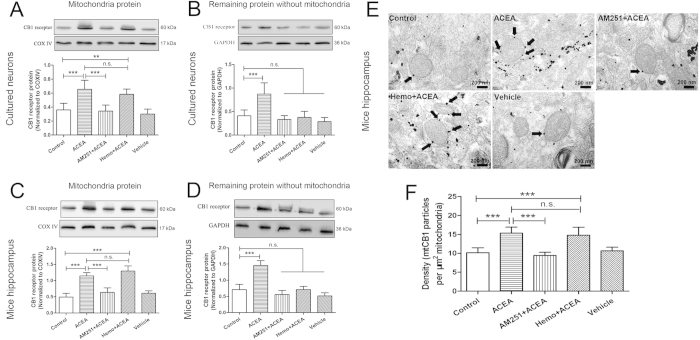
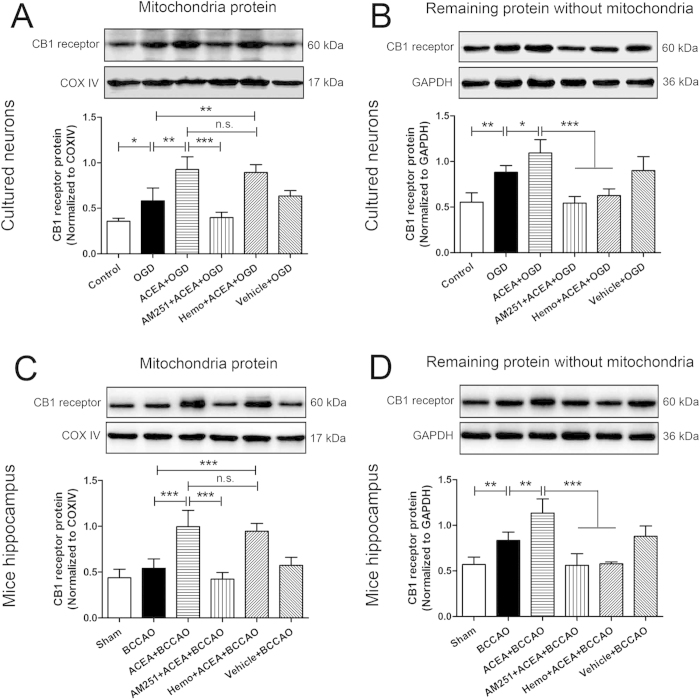
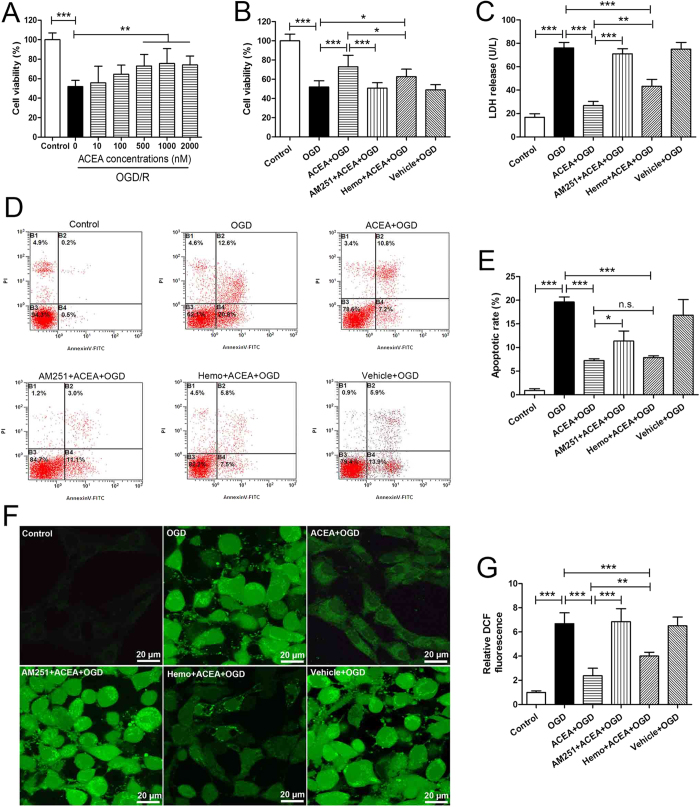
References
-
- Laver S., Farrow C., Turner D. & Nolan J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med 30, 2126–2128 (2004). - PubMed
-
- Nichol G. et al. Regional systems of care for out-of-hospital cardiac arrest: A policy statement from the American Heart Association. Circulation 121, 709–729 (2010). - PubMed
-
- Roine R. O., Kajaste S. & Kaste M. Neuropsychological sequelae of cardiac arrest. JAMA 269, 237–242 (1993). - PubMed
-
- MacAskill A. F. & Kittler J. T. Control of mitochondrial transport and localization in neurons. Trends Cell Biol 20, 102–112 (2010). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
