

Genomic approaches to identifying targets for treating β hemoglobinopathies
- PMID: 26215470
- PMCID: PMC4517356
- DOI: 10.1186/s12920-015-0120-2
Genomic approaches to identifying targets for treating β hemoglobinopathies
Abstract
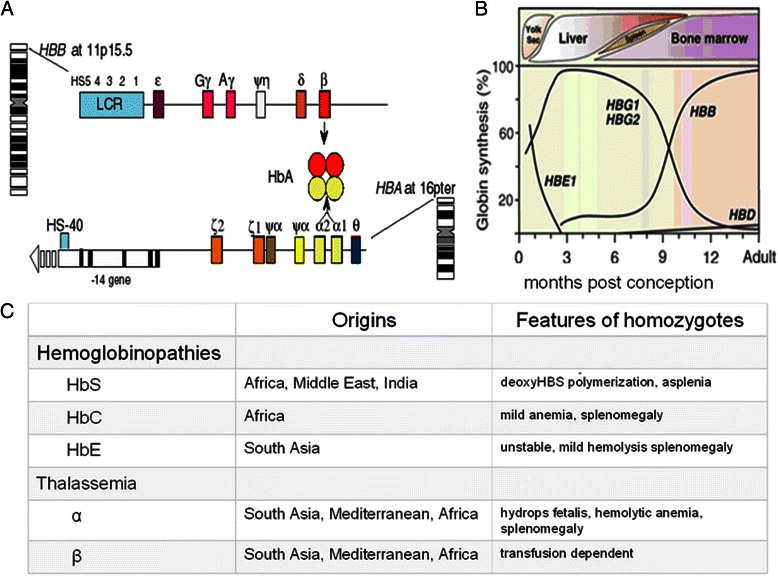
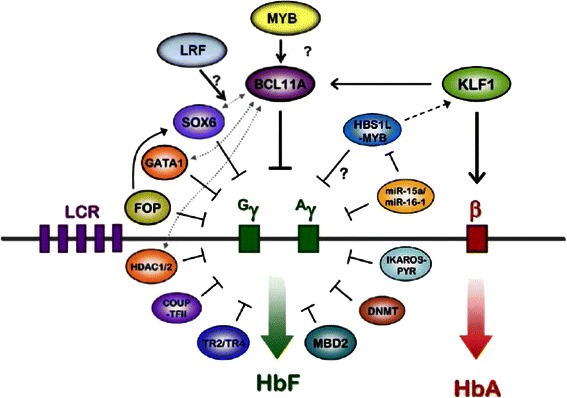
Sickle cell disease and β thalassemia are common severe diseases with little effective pathophysiologically-based treatment. Their phenotypic heterogeneity prompted genomic approaches to identify modifiers that ultimately might be exploited therapeutically. Fetal hemoglobin (HbF) is the major modulator of the phenotype of the β hemoglobinopathies. HbF inhibits deoxyHbS polymerization and in β thalassemia compensates for the reduction of HbA. The major success of genomics has been a better understanding the genetic regulation of HbF by identifying the major quantitative trait loci for this trait. If the targets identified can lead to means of increasing HbF to therapeutic levels in sufficient numbers of sickle or β-thalassemia erythrocytes, the pathophysiology of these diseases would be reversed. The availability of new target loci, high-throughput drug screening, and recent advances in genome editing provide the opportunity for new approaches to therapeutically increasing HbF production.
Figures
References
-
- Organization WH. Management of Haemoglobin Disorders, Report of Joint WHO/TIF Meeting. Nicosia: Cyprus; 2007.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
