

Pharmacological evidence for a role of the transient receptor potential canonical 3 (TRPC3) channel in endoplasmic reticulum stress-induced apoptosis of human coronary artery endothelial cells
- PMID: 26215710
- PMCID: PMC4706806
- DOI: 10.1016/j.vph.2015.07.011
Pharmacological evidence for a role of the transient receptor potential canonical 3 (TRPC3) channel in endoplasmic reticulum stress-induced apoptosis of human coronary artery endothelial cells
Abstract
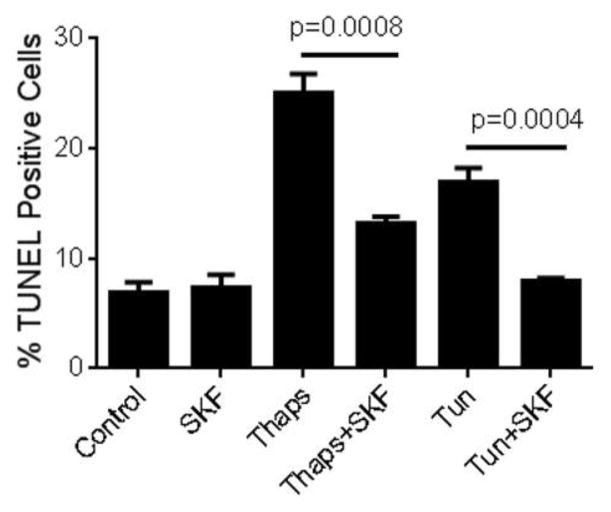
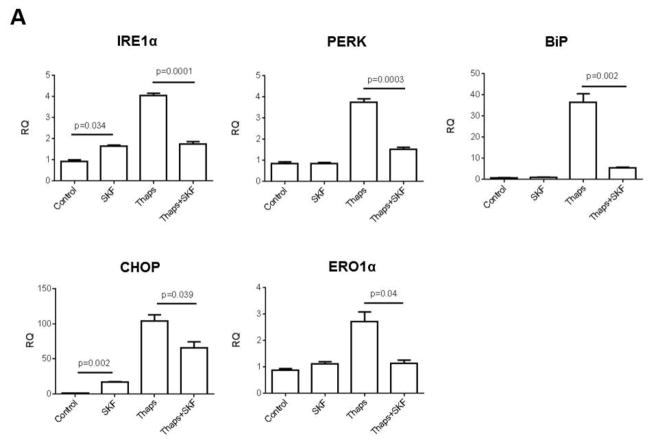
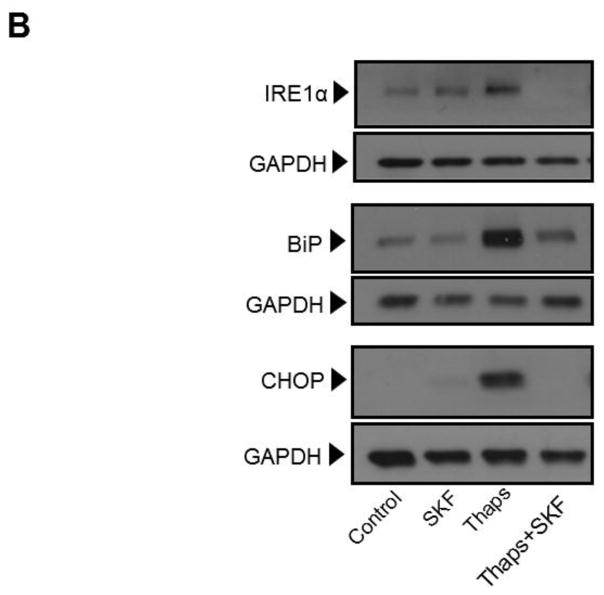
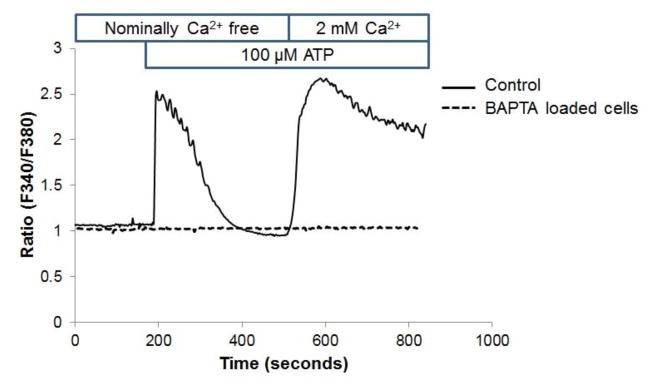
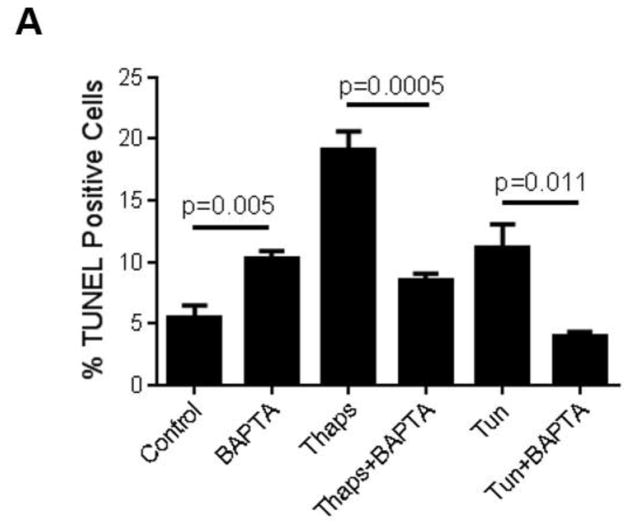
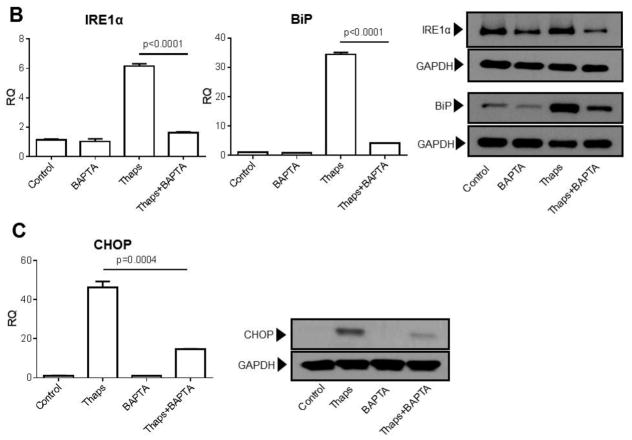
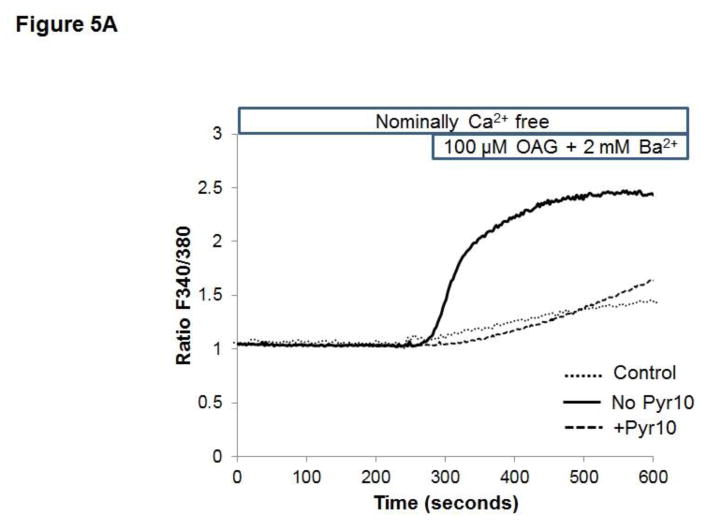
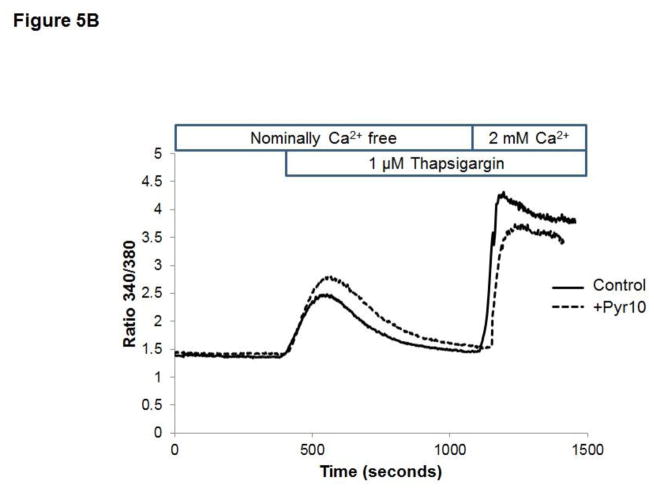
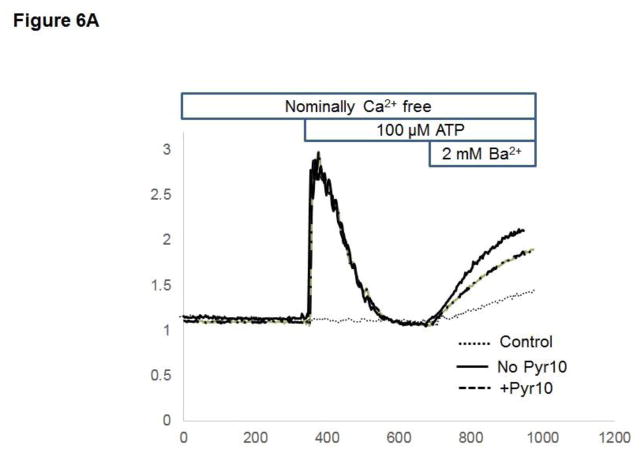
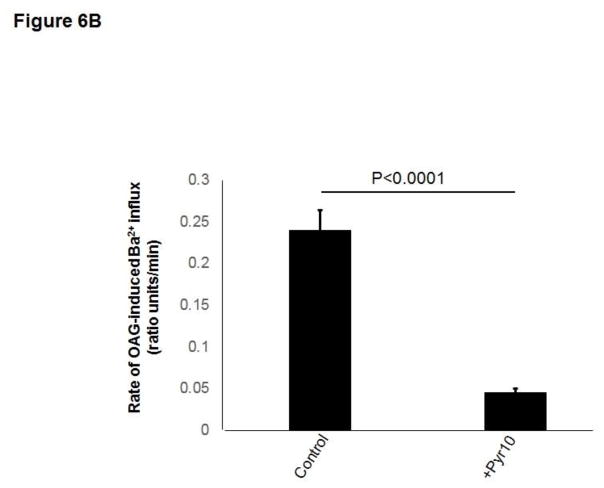
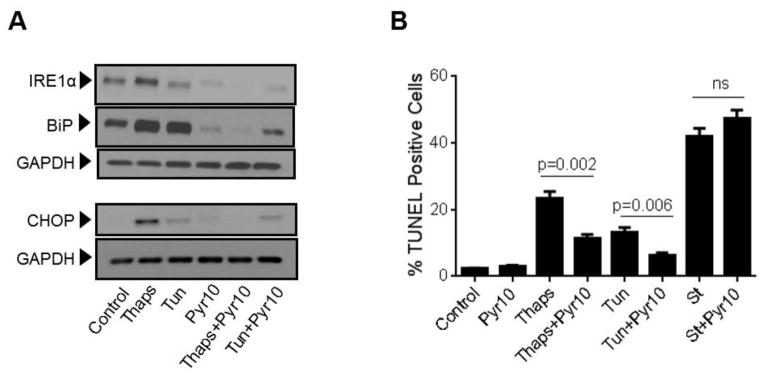
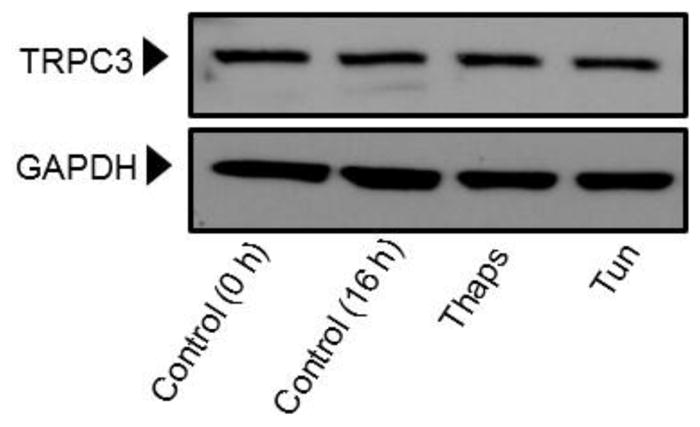
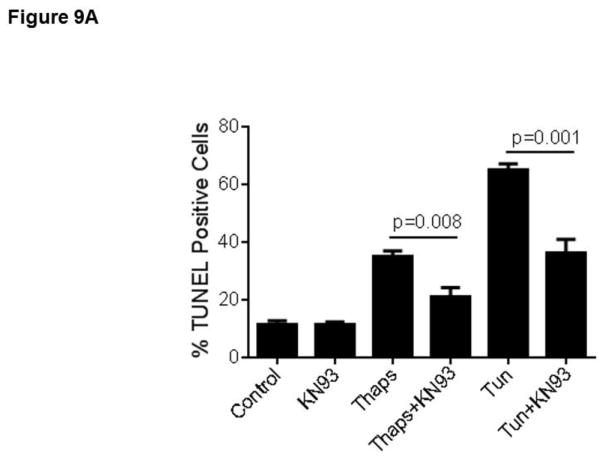
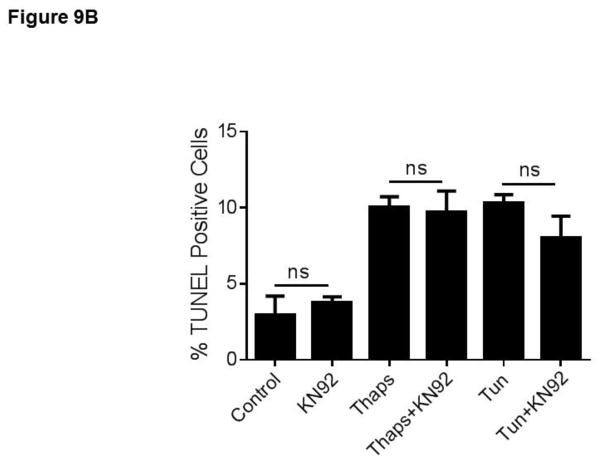
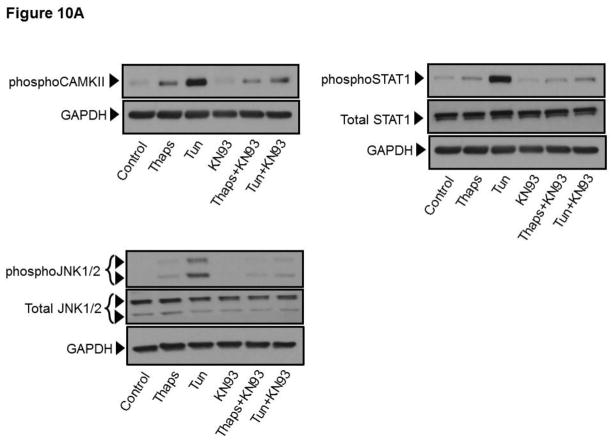
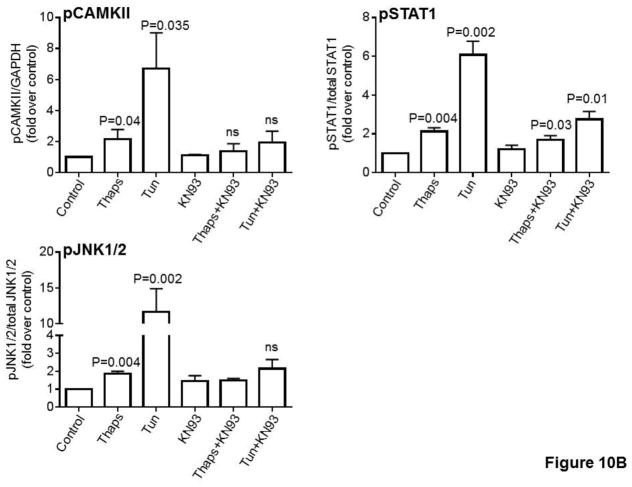
Unresolved endoplasmic reticulum (ER) stress, with the subsequent persistent activation of the unfolded protein response (UPR) is a well-recognized mechanism of endothelial cell apoptosis with a major impact on the integrity of the endothelium during the course of cardiovascular diseases. As in other cell types, Ca(2+) influx into endothelial cells can promote ER stress and/or contribute to mechanisms associated with it. In previous work we showed that in human coronary artery endothelial cells (HCAECs) the Ca(2+)-permeable non-selective cation channel Transient Receptor Potential Canonical 3 (TRPC3) mediates constitutive Ca(2+) influx which is critical for operation of inflammatory signaling in these cells, through a mechanism that entails coupling of TRPC3 constitutive function to activation of Ca(2+)/calmodulin-dependent protein kinase II (CAMKII). TRPC3 has been linked to UPR signaling and apoptosis in cells other than endothelial, and CAMKII is a mediator of ER stress-induced apoptosis in various cell types, including endothelial cells. In the present work we used a pharmacological approach to examine whether in HCAECs TRPC3 and CAMKII also contribute to mechanisms of ER stress-induced apoptosis. The findings show for the first time that in HCAECs activation of the UPR and the subsequent ER stress-induced apoptosis exhibit a strong requirement for constitutive Ca(2+) influx and that TRPC3 contributes to this process. In addition, we obtained evidence indicating that, similar to its roles in non-endothelial cells, CAMKII participates in ER stress-induced apoptosis in HCAECs.
Keywords: Endoplasmic reticulum stress; Endothelial cell apoptosis; TRPC3 channels.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures

Similar articles
-
The constitutive function of native TRPC3 channels modulates vascular cell adhesion molecule-1 expression in coronary endothelial cells through nuclear factor kappaB signaling.Circ Res. 2010 May 14;106(9):1479-88. doi: 10.1161/CIRCRESAHA.109.213314. Epub 2010 Apr 1. Circ Res. 2010. PMID: 20360250
-
Reduced endoplasmic reticulum stress-induced apoptosis and impaired unfolded protein response in TRPC3-deficient M1 macrophages.Am J Physiol Cell Physiol. 2014 Sep 15;307(6):C521-31. doi: 10.1152/ajpcell.00369.2013. Epub 2014 Jul 16. Am J Physiol Cell Physiol. 2014. PMID: 25031020 Free PMC article.
-
Involvement of native TRPC3 proteins in ATP-dependent expression of VCAM-1 and monocyte adherence in coronary artery endothelial cells.Arterioscler Thromb Vasc Biol. 2008 Nov;28(11):2049-55. doi: 10.1161/ATVBAHA.108.175356. Epub 2008 Sep 11. Arterioscler Thromb Vasc Biol. 2008. PMID: 18787184
-
Transient receptor potential canonical type 3 channels: Interactions, role and relevance - A vascular focus.Pharmacol Ther. 2017 Jun;174:79-96. doi: 10.1016/j.pharmthera.2017.02.022. Epub 2017 Feb 14. Pharmacol Ther. 2017. PMID: 28223224 Review.
-
On the role of endothelial TRPC3 channels in endothelial dysfunction and cardiovascular disease.Cardiovasc Hematol Agents Med Chem. 2012 Sep;10(3):265-74. doi: 10.2174/187152512802651051. Cardiovasc Hematol Agents Med Chem. 2012. PMID: 22827251 Free PMC article. Review.
Cited by
-
Six Decades of History of Hypertension Research at the University of Toledo: Highlighting Pioneering Contributions in Biochemistry, Genetics, and Host-Microbiota Interactions.Curr Hypertens Rep. 2022 Dec;24(12):669-685. doi: 10.1007/s11906-022-01226-0. Epub 2022 Oct 27. Curr Hypertens Rep. 2022. PMID: 36301488 Free PMC article. Review.
-
TRPC channels: Structure, function, regulation and recent advances in small molecular probes.Pharmacol Ther. 2020 May;209:107497. doi: 10.1016/j.pharmthera.2020.107497. Epub 2020 Jan 28. Pharmacol Ther. 2020. PMID: 32004513 Free PMC article. Review.
-
miR-454-3p prevents ox-LDL-induced apoptosis in HAECs by targeting TRPC3.Exp Ther Med. 2021 Apr;21(4):323. doi: 10.3892/etm.2021.9754. Epub 2021 Feb 5. Exp Ther Med. 2021. PMID: 33732296 Free PMC article.
-
Expression of Oxidative Stress and Inflammatory Indicators for Coronary Artery Disease in Kawasaki Disease.Mediterr J Hematol Infect Dis. 2024 Jul 1;16(1):e2024052. doi: 10.4084/MJHID.2024.052. eCollection 2024. Mediterr J Hematol Infect Dis. 2024. PMID: 38984102 Free PMC article.
-
Early steatohepatitis in hyperlipidemic mice with endothelial-specific gain of TRPC3 function precedes changes in aortic atherosclerosis.Physiol Genomics. 2016 Aug 1;48(8):644-9. doi: 10.1152/physiolgenomics.00067.2016. Epub 2016 Jul 22. Physiol Genomics. 2016. PMID: 27449657 Free PMC article.
References
-
- Baldi CVG, Boland R. Capacitative calcium influx in human epithelial breast cancer and non-tumorigenic cells occurs through Ca2+ entry pathways with different permeabilities to divalent cations. Journal of cellular biochemistry. 2003;88:1265–1272. - PubMed
-
- Chen S, He FF, Wang H, Fang Z, Shao N, Tian XJ, Liu JS, Zhu ZH, Wang YM, Wang S, Huang K, Zhang C. Calcium entry via TRPC6 mediates albumin overload-induced endoplasmic reticulum stress and apoptosis in podocytes. Cell calcium. 2011;50:523–529. - PubMed
-
- Hollien J. Evolution of the unfolded protein response. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2013;1833:2458–2463. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
