

APOL1 Kidney Disease Risk Variants: An Evolving Landscape
- PMID: 26215860
- PMCID: PMC4562465
- DOI: 10.1016/j.semnephrol.2015.04.008
APOL1 Kidney Disease Risk Variants: An Evolving Landscape
Abstract
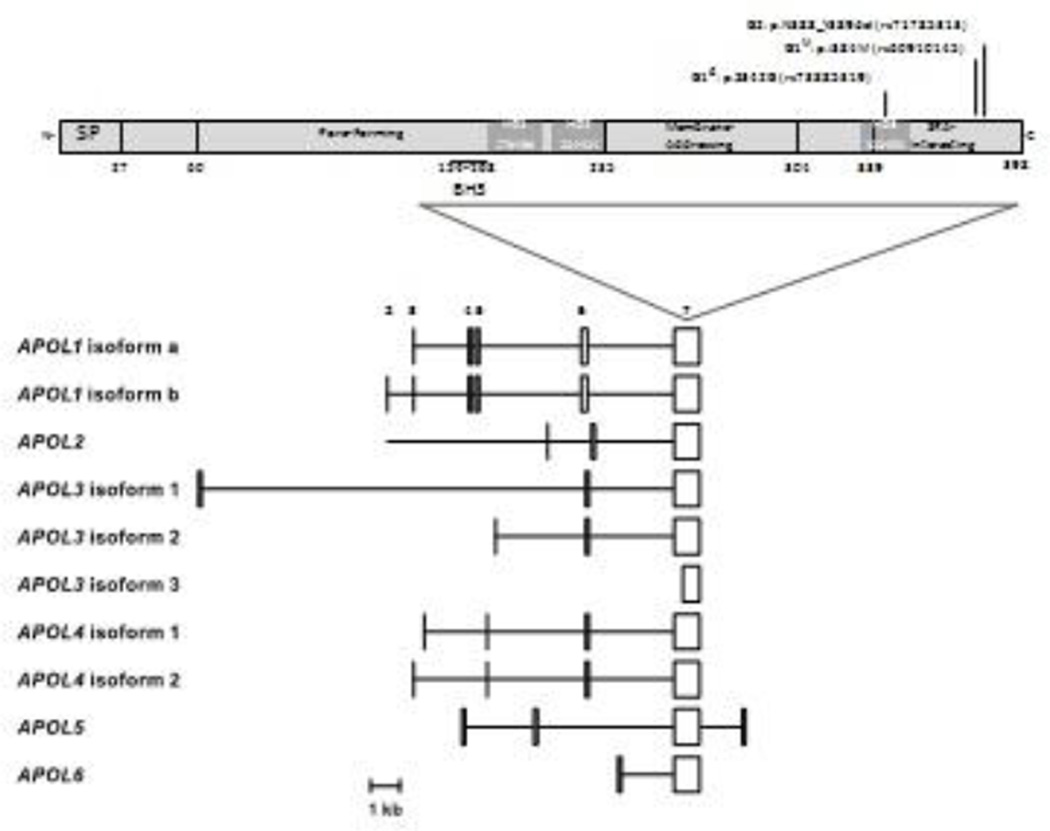
Apolipoprotein L1 (APOL1) genetic variants account for much of the excess risk of chronic and end-stage kidney disease, which results in a significant global health disparity for persons of African ancestry. We estimate the lifetime risk of kidney disease in APOL1 dual-risk allele individuals to be at least 15%. Experimental evidence suggests a direct role of APOL1 in pore formation, cellular injury, and programmed cell death in renal injury. The APOL1 BH3 motif, often associated with cell death, is unlikely to play a role in APOL1-induced cytotoxicity because it is not conserved within the APOL family and is dispensable for cell death in vitro. We discuss two models for APOL1 trypanolytic activity: one involving lysosome permeabilization and another involving colloid-osmotic swelling of the cell body, as well as their relevance to human pathophysiology. Experimental evidence from human cell culture models suggests that both mechanisms may be operative. A systems biology approach whereby APOL1-associated perturbations in gene and protein expression in affected individuals are correlated with molecular pathways may be productive to elucidate APOL1 function in vivo.
Keywords: APOL1; Health disparities; chronic kidney disease; focal segmental glomerulosclerosis; innate immunity.
Published by Elsevier Inc.
Conflict of interest statement
conflict of interest: No conflicts of interest.
Figures



References
-
- United States Renal Data System. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2014. Annual Data Report: Epidemiology of Kidney Disease in the United States.
-
- Kiberd BA. Cumulative Risk for Developing End-Stage Renal Disease in the US Population. Journal of the American Society of Nephrology. 2002;13:1635–1644. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
