

Functional IRF3 deficiency in a patient with herpes simplex encephalitis
- PMID: 26216125
- PMCID: PMC4548062
- DOI: 10.1084/jem.20142274
Functional IRF3 deficiency in a patient with herpes simplex encephalitis
Abstract
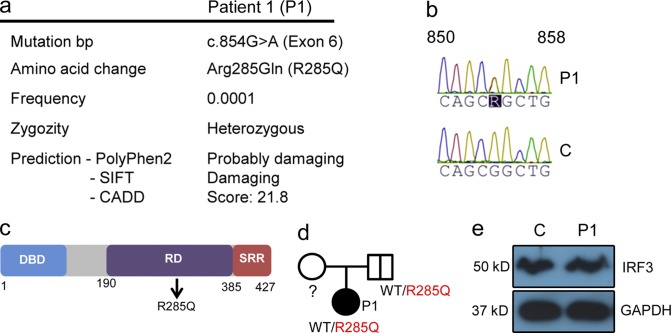
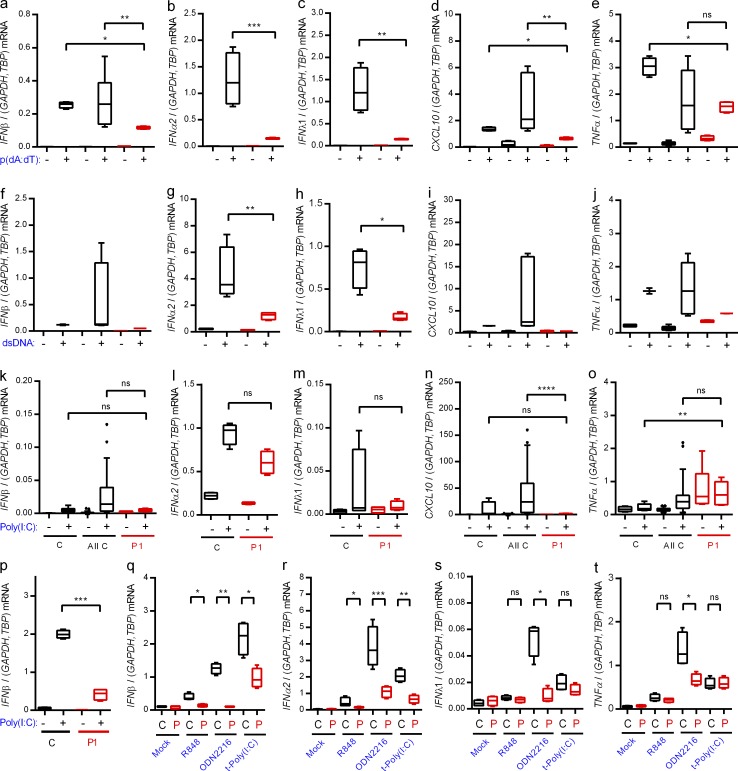
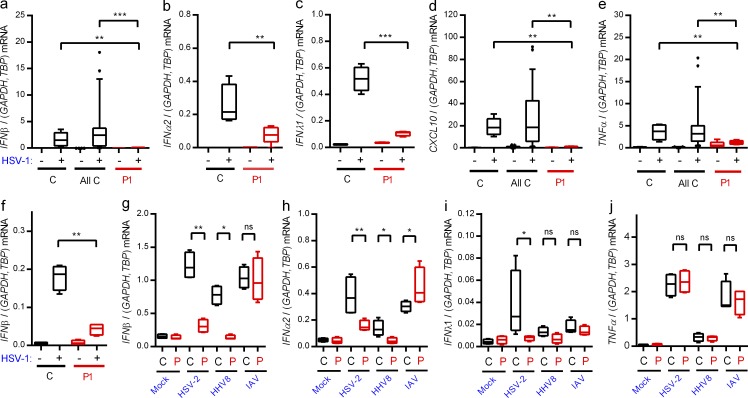
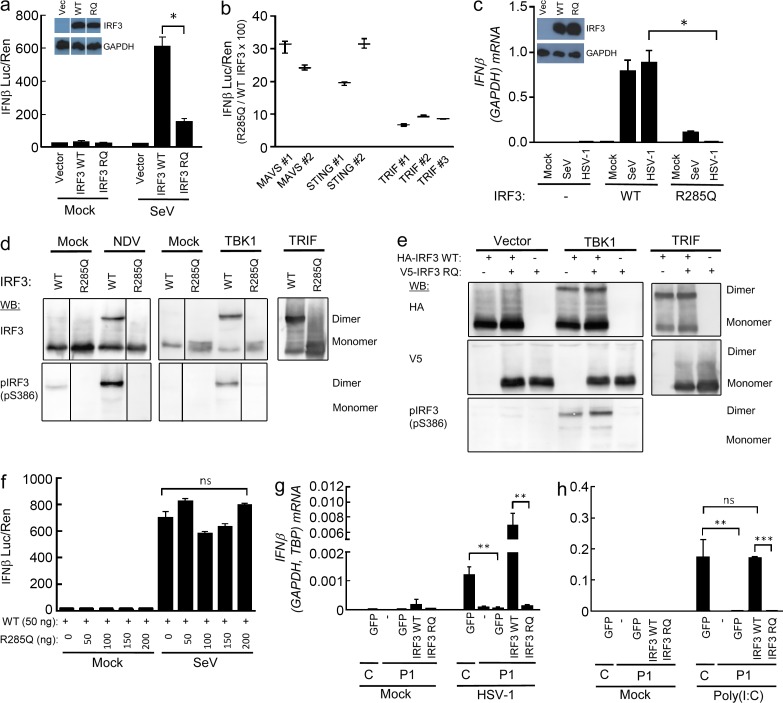
Herpes simplex encephalitis (HSE) in children has previously been linked to defects in type I interferon (IFN) production downstream of Toll-like receptor 3. Here, we describe a novel genetic etiology of HSE by identifying a heterozygous loss-of-function mutation in the IFN regulatory factor 3 (IRF3) gene, leading to autosomal dominant (AD) IRF3 deficiency by haploinsufficiency, in an adolescent female patient with HSE. IRF3 is activated by most pattern recognition receptors recognizing viral infections and plays an essential role in induction of type I IFN. The identified IRF3 R285Q amino acid substitution results in impaired IFN responses to HSV-1 infection and particularly impairs signaling through the TLR3-TRIF pathway. In addition, the R285Q mutant of IRF3 fails to become phosphorylated at S386 and undergo dimerization, and thus has impaired ability to activate transcription. Finally, transduction with WT IRF3 rescues the ability of patient fibroblasts to express IFN in response to HSV-1 infection. The identification of IRF3 deficiency in HSE provides the first description of a defect in an IFN-regulating transcription factor conferring increased susceptibility to a viral infection in the CNS in humans.
© 2015 Andersen et al.
Figures
Comment in
-
Inborn errors underlying herpes simplex encephalitis: From TLR3 to IRF3.J Exp Med. 2015 Aug 24;212(9):1342-3. doi: 10.1084/jem.2129insight4. J Exp Med. 2015. PMID: 26304982 Free PMC article. No abstract available.
References
-
- Abel L., Plancoulaine S., Jouanguy E., Zhang S.Y., Mahfoufi N., Nicolas N., Sancho-Shimizu V., Alcaïs A., Guo Y., Cardon A., et al. . 2010. Age-dependent Mendelian predisposition to herpes simplex virus type 1 encephalitis in childhood. J. Pediatr. 157:623–629: e1. 10.1016/j.jpeds.2010.04.020 - DOI - PubMed
-
- Audry M., Ciancanelli M., Yang K., Cobat A., Chang H.H., Sancho-Shimizu V., Lorenzo L., Niehues T., Reichenbach J., Li X.X., et al. . 2011. NEMO is a key component of NF-κB- and IRF-3-dependent TLR3-mediated immunity to herpes simplex virus. J. Allergy Clin. Immunol. 128:610–617: e1–e4. 10.1016/j.jaci.2011.04.059 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
