

Roles of unphosphorylated ISGF3 in HCV infection and interferon responsiveness
- PMID: 26216956
- PMCID: PMC4547285
- DOI: 10.1073/pnas.1513341112
Roles of unphosphorylated ISGF3 in HCV infection and interferon responsiveness
Abstract
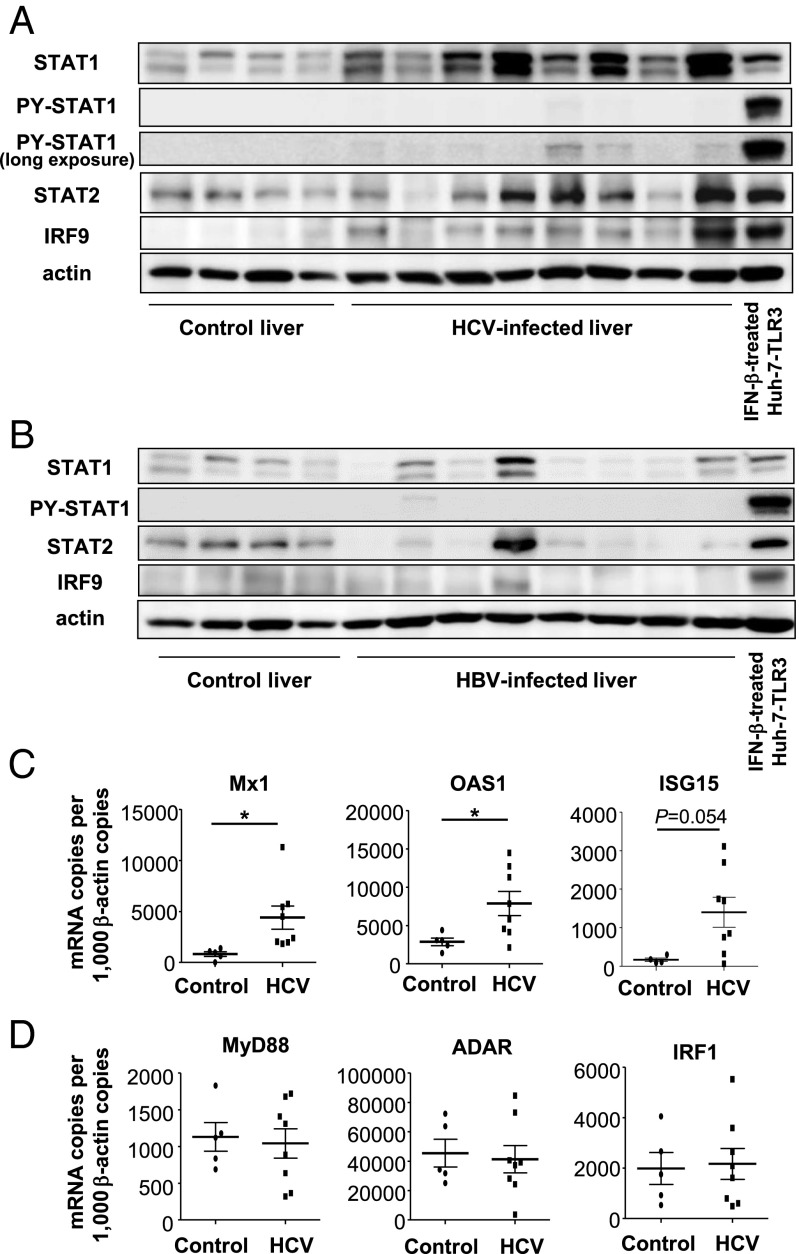
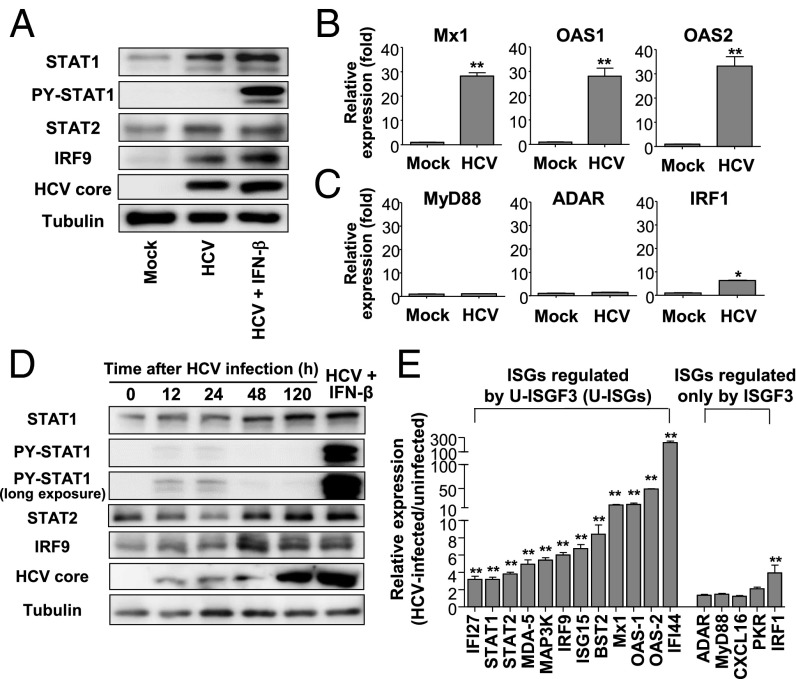
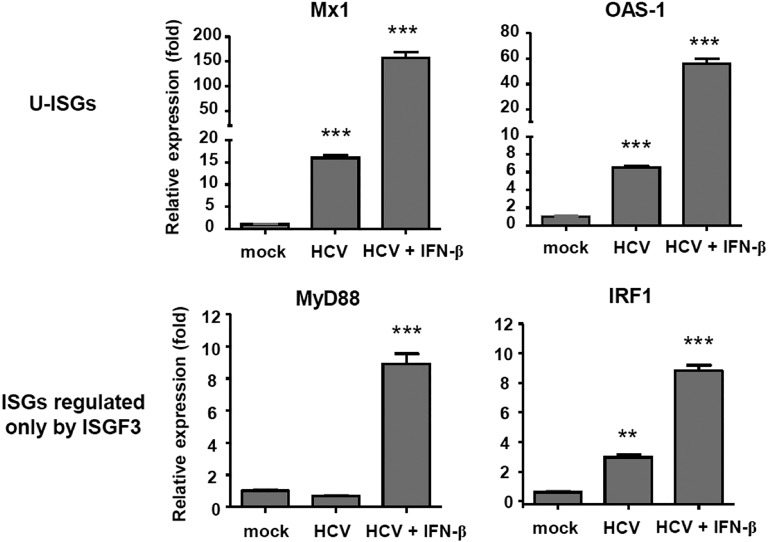
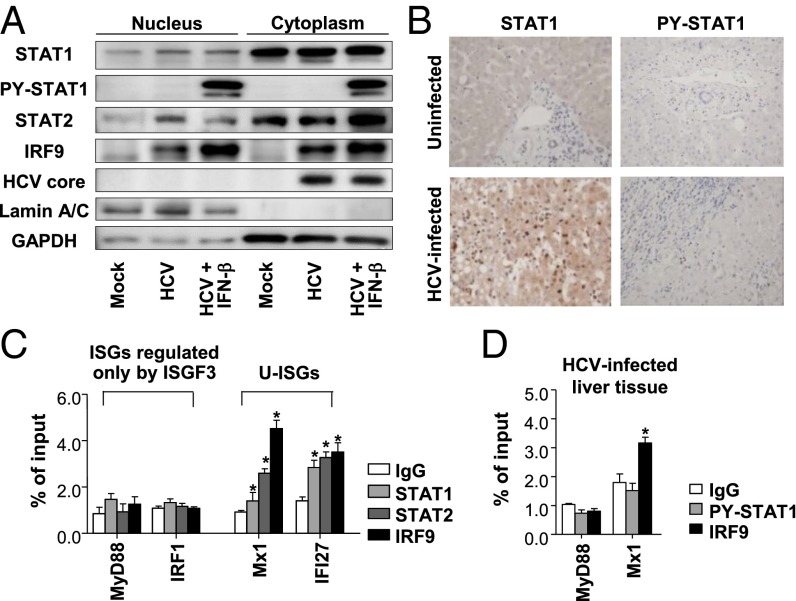
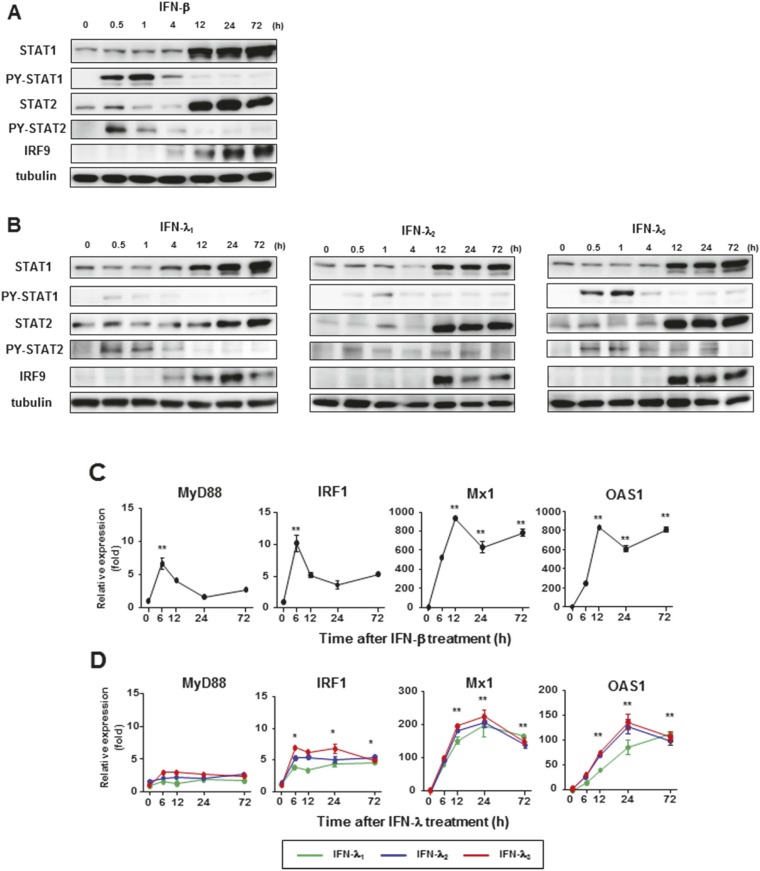
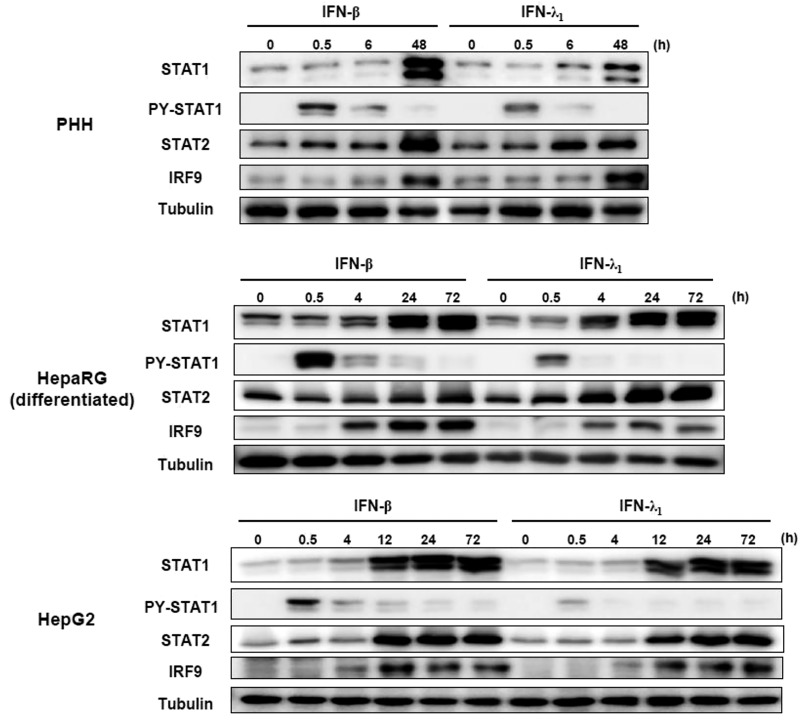
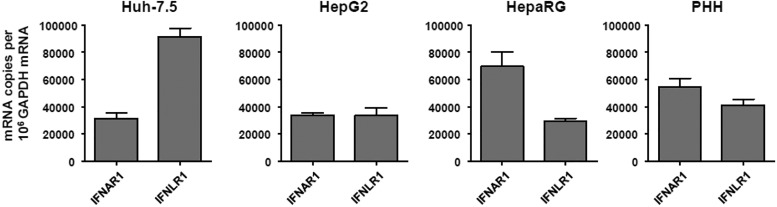
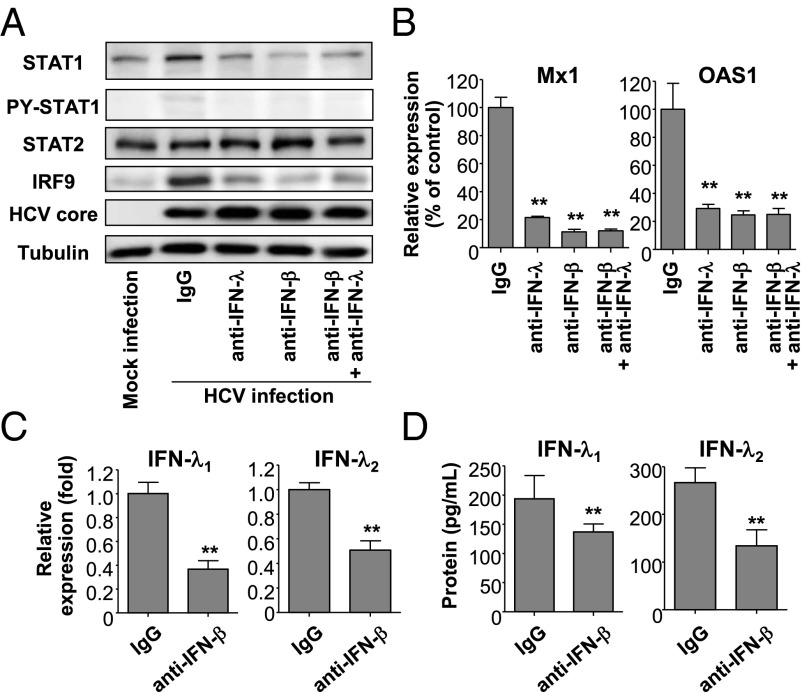
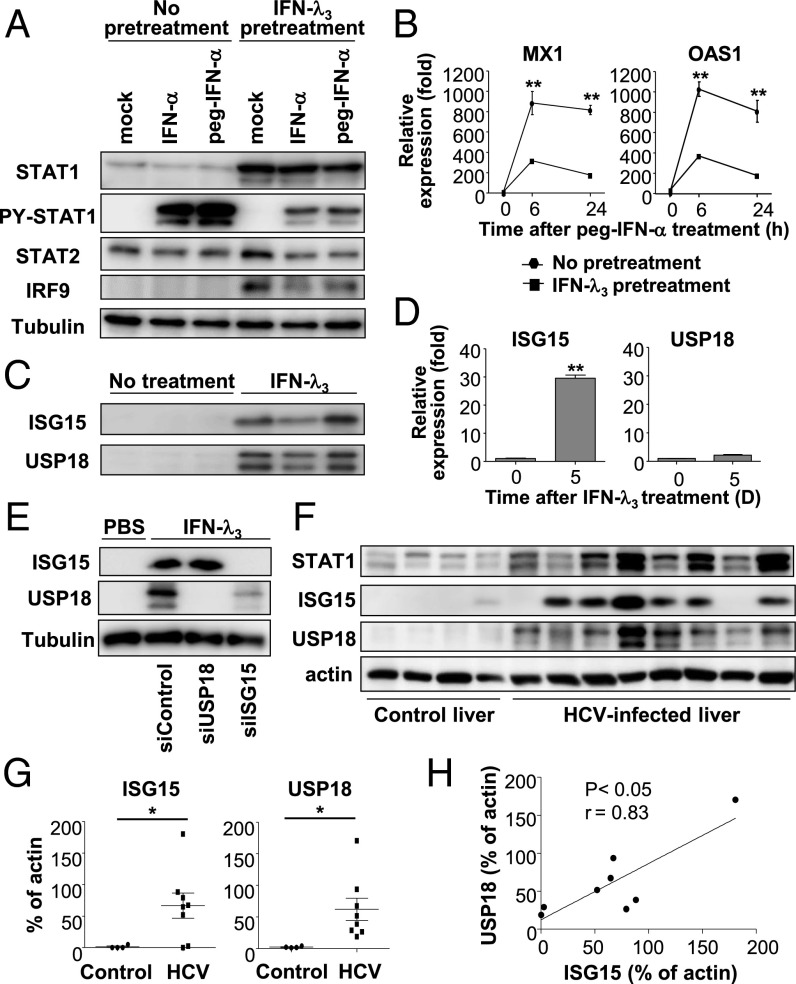
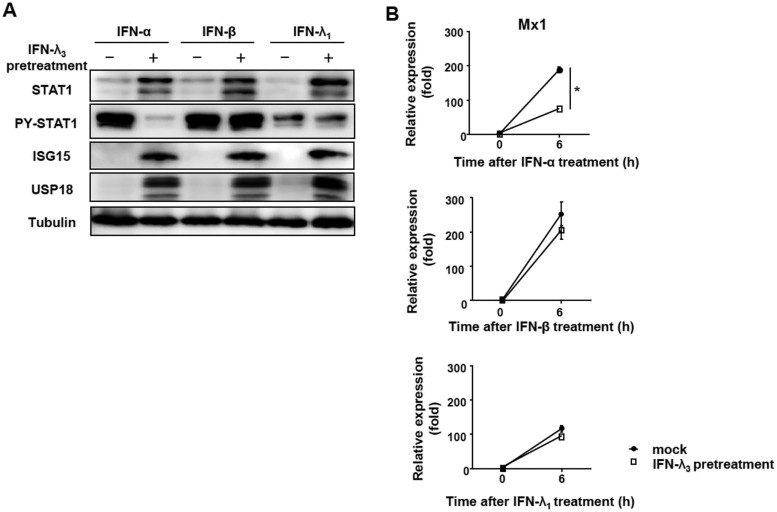
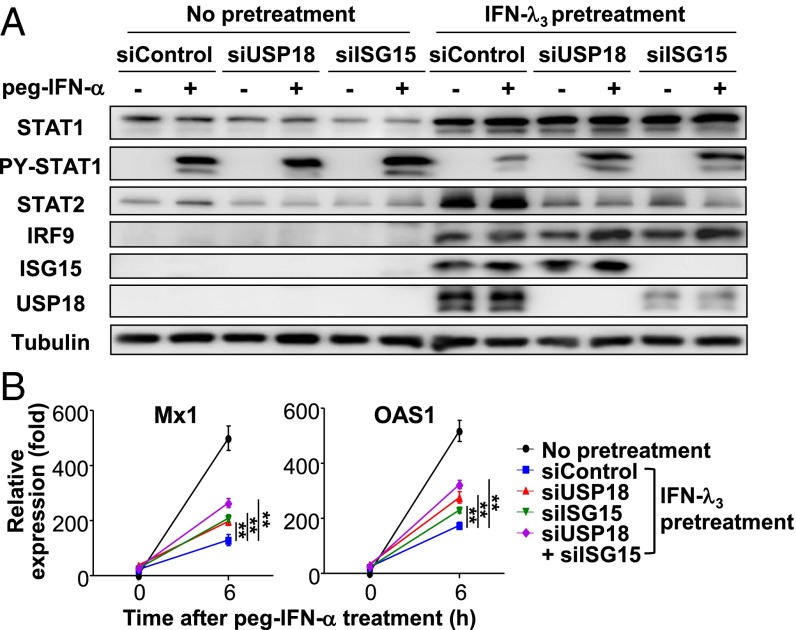
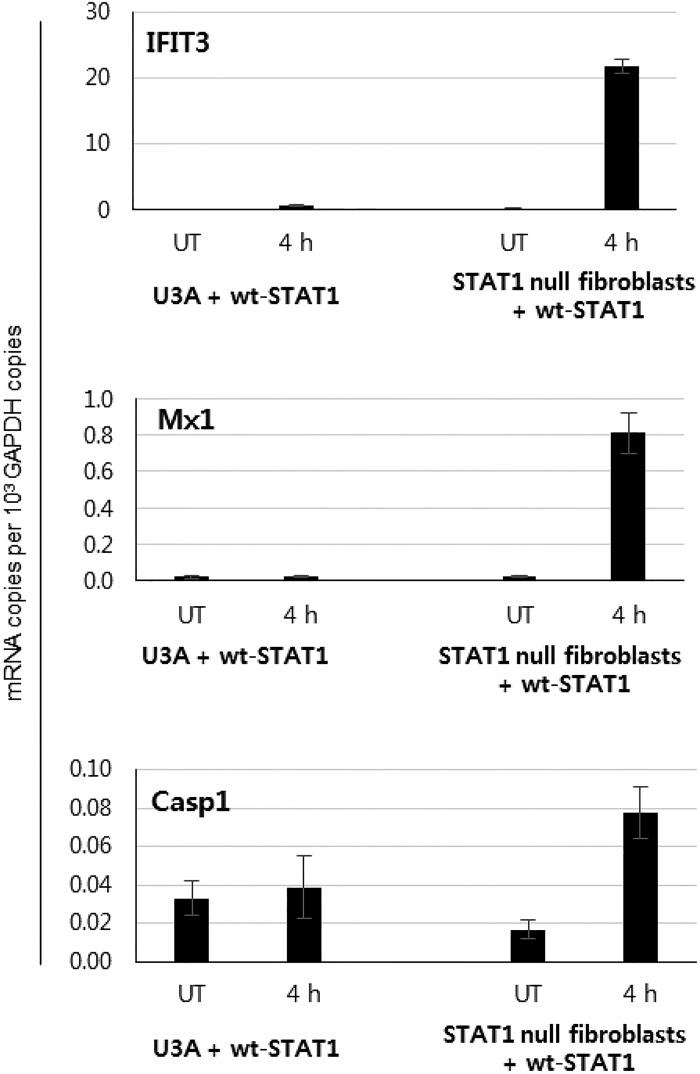
Up-regulation of IFN-stimulated genes (ISGs) is sustained in hepatitis C virus (HCV)-infected livers. Here, we investigated the mechanism of prolonged ISG expression and its role in IFN responsiveness during HCV infection in relation to unphosphorylated IFN-stimulated gene factor 3 (U-ISGF3), recently identified as a tripartite transcription factor formed by high levels of IFN response factor 9 (IRF9), STAT1, and STAT2 without tyrosine phosphorylation of the STATs. The level of U-ISGF3, but not tyrosine phosphorylated STAT1, is significantly elevated in response to IFN-λ and IFN-β during chronic HCV infection. U-ISGF3 prolongs the expression of a subset of ISGs and restricts HCV chronic replication. However, paradoxically, high levels of U-ISGF3 also confer unresponsiveness to IFN-α therapy. As a mechanism of U-ISGF3-induced resistance to IFN-α, we found that ISG15, a U-ISGF3-induced protein, sustains the abundance of ubiquitin-specific protease 18 (USP18), a negative regulator of IFN signaling. Our data demonstrate that U-ISGF3 induced by IFN-λs and -β drives prolonged expression of a set of ISGs, leading to chronic activation of innate responses and conferring a lack of response to IFN-α in HCV-infected liver.
Keywords: U-ISGF3; hepatitis C virus; interferon; interferon-stimulated genes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
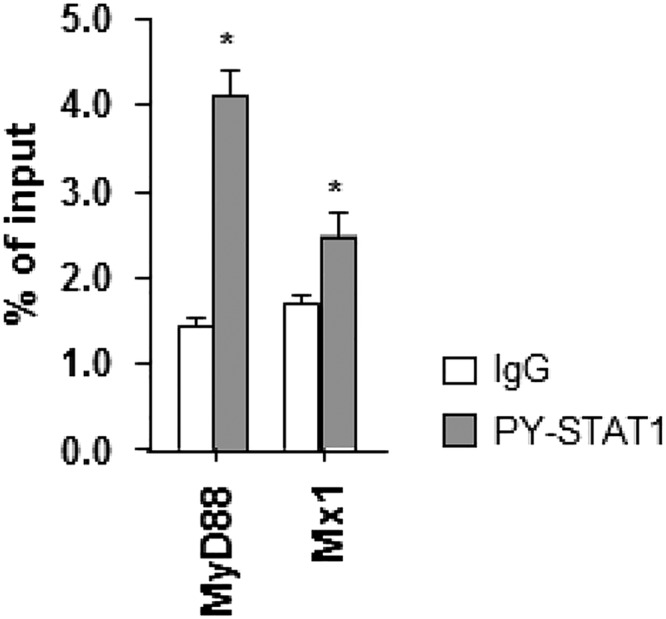
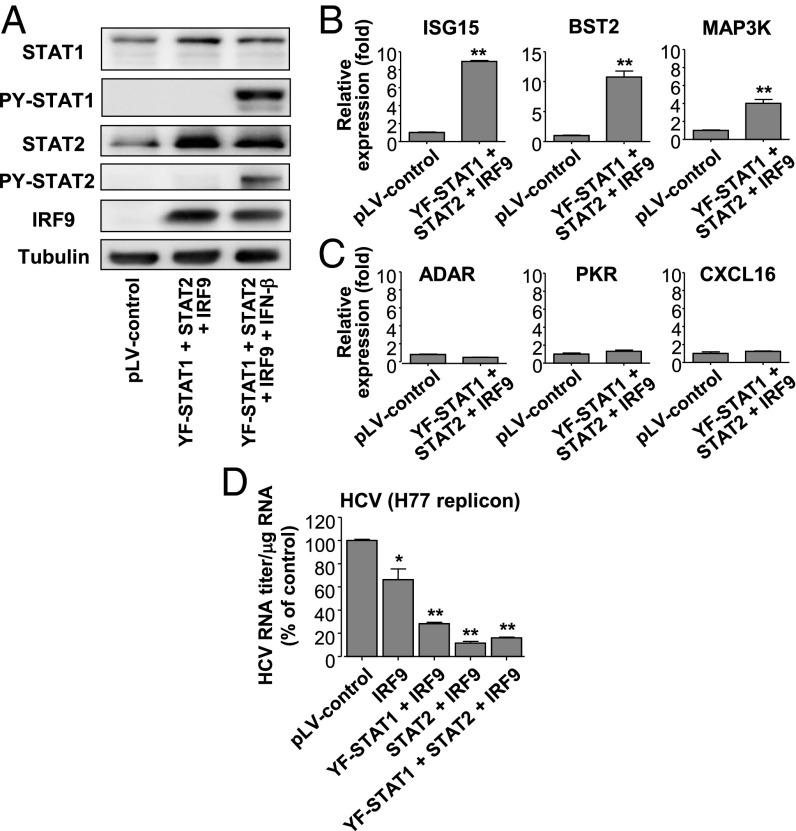
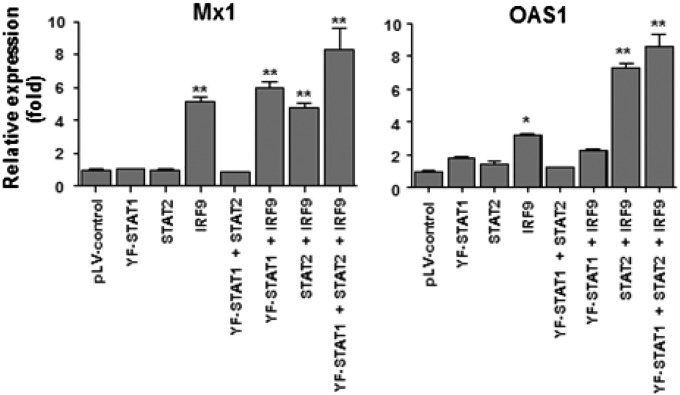
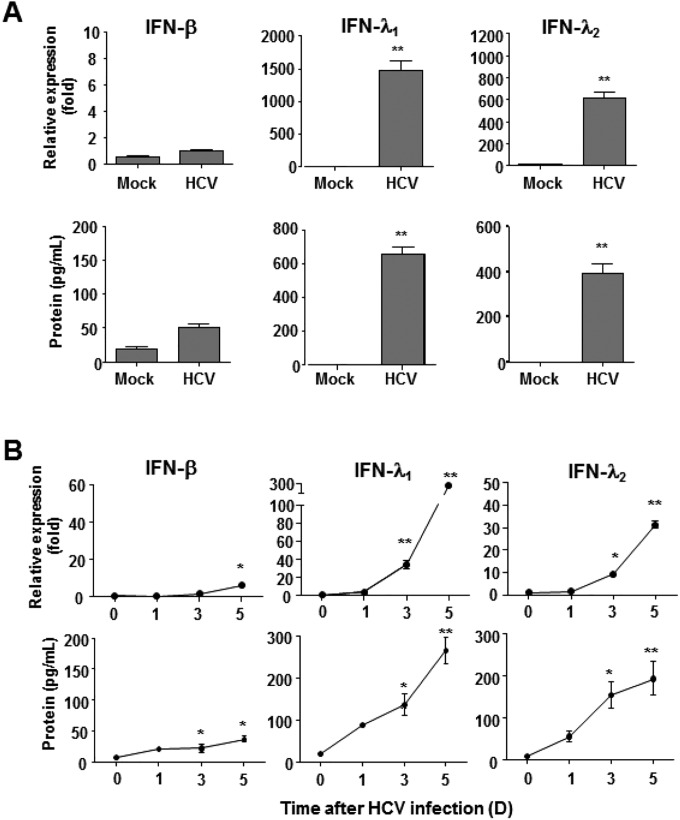
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
