

Mitochondrial calcium overload is a key determinant in heart failure
- PMID: 26217001
- PMCID: PMC4568687
- DOI: 10.1073/pnas.1513047112
Mitochondrial calcium overload is a key determinant in heart failure
Abstract
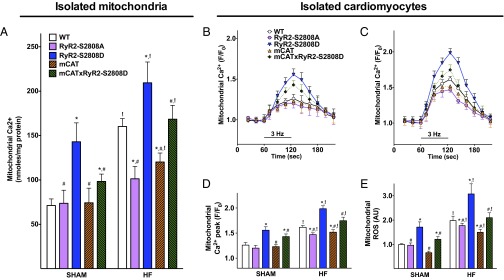
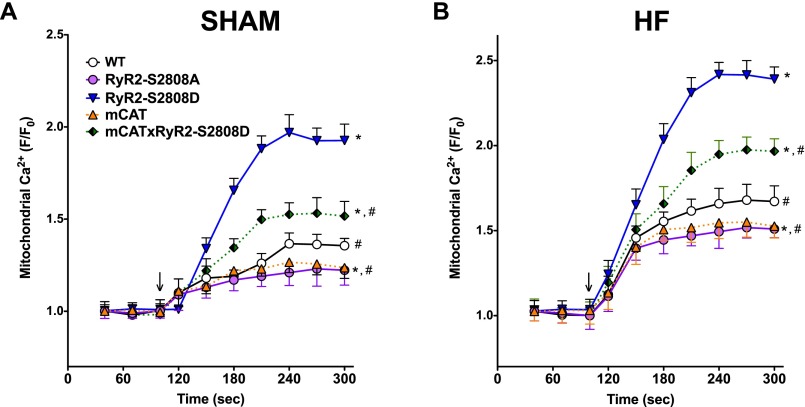
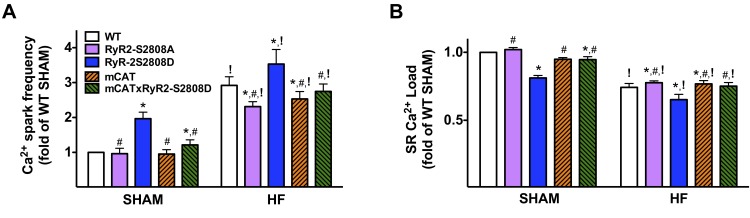
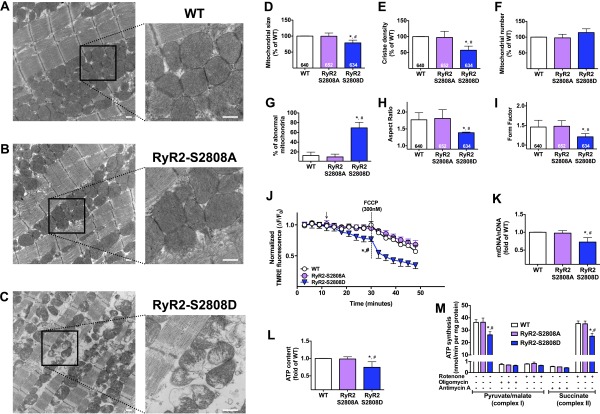
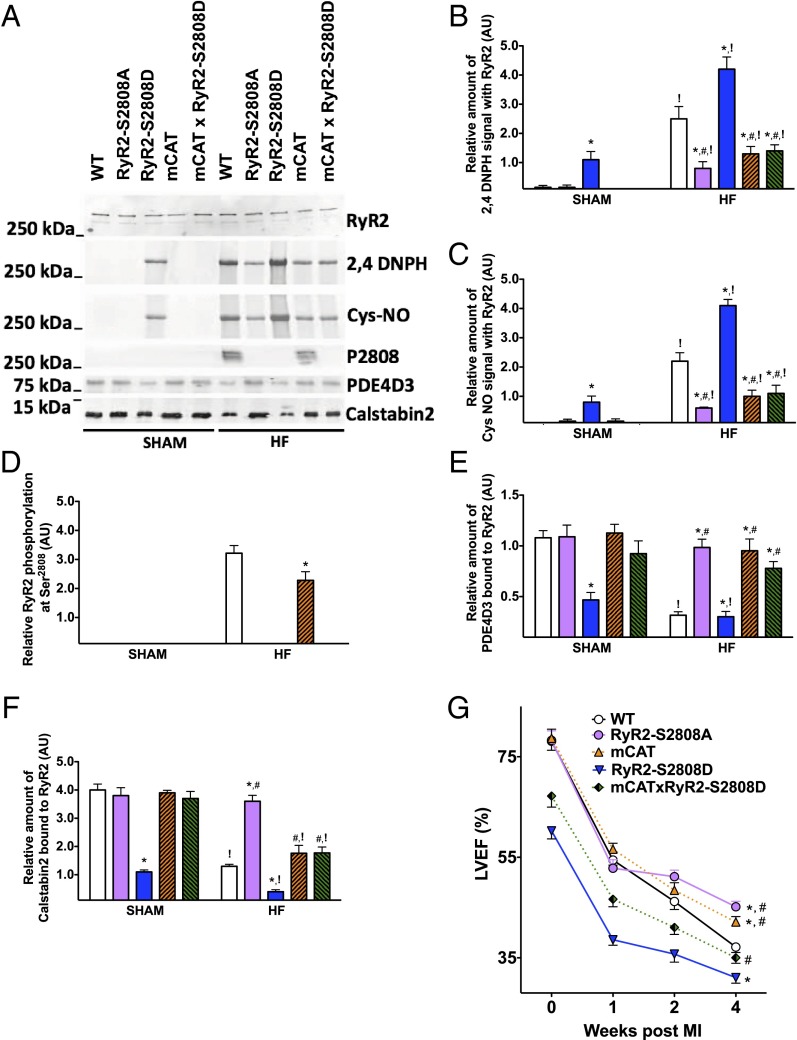
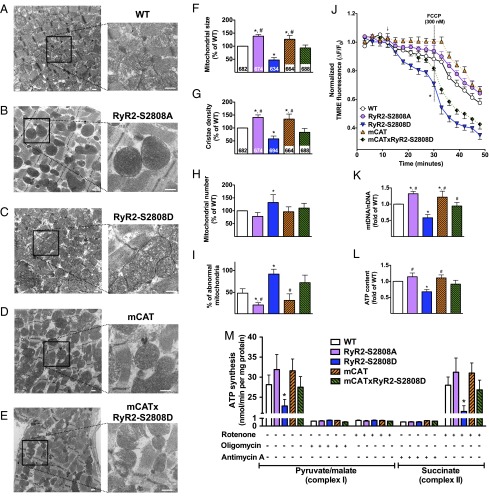
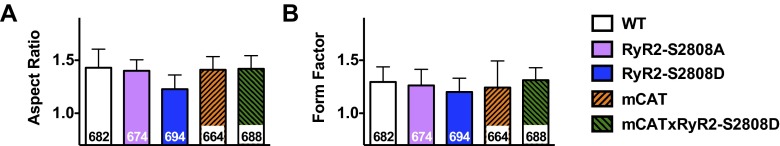
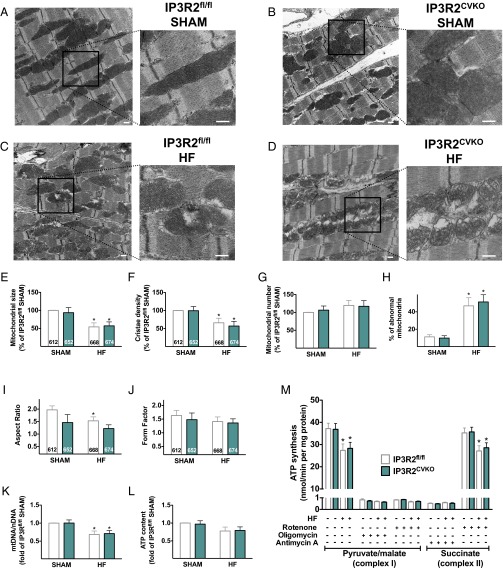
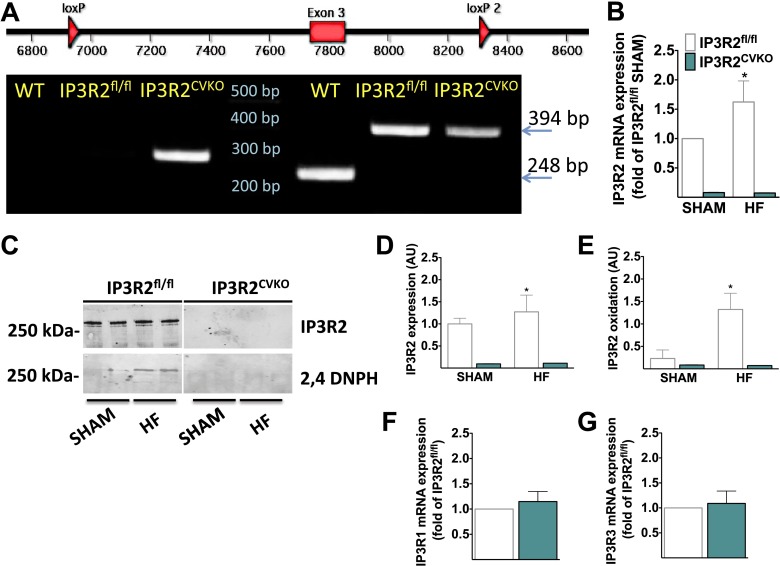
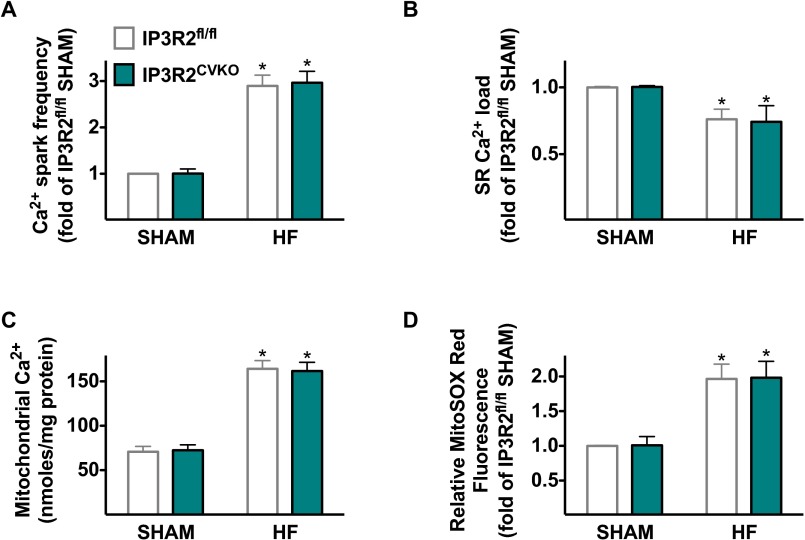
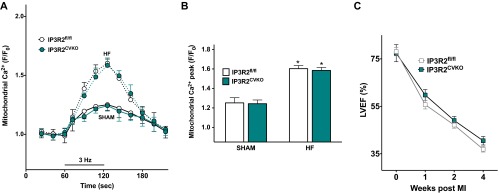
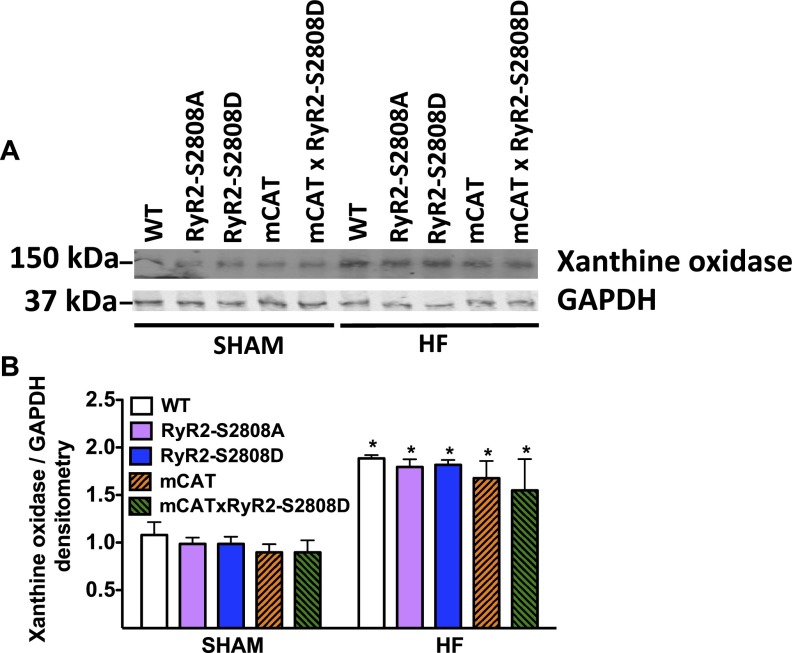
Calcium (Ca2+) released from the sarcoplasmic reticulum (SR) is crucial for excitation-contraction (E-C) coupling. Mitochondria, the major source of energy, in the form of ATP, required for cardiac contractility, are closely interconnected with the SR, and Ca2+ is essential for optimal function of these organelles. However, Ca2+ accumulation can impair mitochondrial function, leading to reduced ATP production and increased release of reactive oxygen species (ROS). Oxidative stress contributes to heart failure (HF), but whether mitochondrial Ca2+ plays a mechanistic role in HF remains unresolved. Here, we show for the first time, to our knowledge, that diastolic SR Ca2+ leak causes mitochondrial Ca2+ overload and dysfunction in a murine model of postmyocardial infarction HF. There are two forms of Ca2+ release channels on cardiac SR: type 2 ryanodine receptors (RyR2s) and type 2 inositol 1,4,5-trisphosphate receptors (IP3R2s). Using murine models harboring RyR2 mutations that either cause or inhibit SR Ca2+ leak, we found that leaky RyR2 channels result in mitochondrial Ca2+ overload, dysmorphology, and malfunction. In contrast, cardiac-specific deletion of IP3R2 had no major effect on mitochondrial fitness in HF. Moreover, genetic enhancement of mitochondrial antioxidant activity improved mitochondrial function and reduced posttranslational modifications of RyR2 macromolecular complex. Our data demonstrate that leaky RyR2, but not IP3R2, channels cause mitochondrial Ca2+ overload and dysfunction in HF.
Keywords: IP3 receptor; calcium; heart failure; mitochondria; ryanodine receptor.
Conflict of interest statement
Conflict of interest statement: A.R.M. is a consultant and member of the board of ARMGO, which is targeting RyR channels for therapeutic purposes.
Figures
Comment in
-
The growing importance of mitochondrial calcium in health and disease.Proc Natl Acad Sci U S A. 2015 Sep 8;112(36):11150-1. doi: 10.1073/pnas.1514284112. Epub 2015 Aug 26. Proc Natl Acad Sci U S A. 2015. PMID: 26311848 Free PMC article. No abstract available.
References
-
- Santulli G, Marks AR. Essential roles of intracellular calcium release channels in muscle, brain, metabolism, and aging. Curr Mol Pharmacol. 2015 in press. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
