

Peptide-Centric Proteome Analysis: An Alternative Strategy for the Analysis of Tandem Mass Spectrometry Data
- PMID: 26217018
- PMCID: PMC4563716
- DOI: 10.1074/mcp.O114.047035
Peptide-Centric Proteome Analysis: An Alternative Strategy for the Analysis of Tandem Mass Spectrometry Data
Abstract
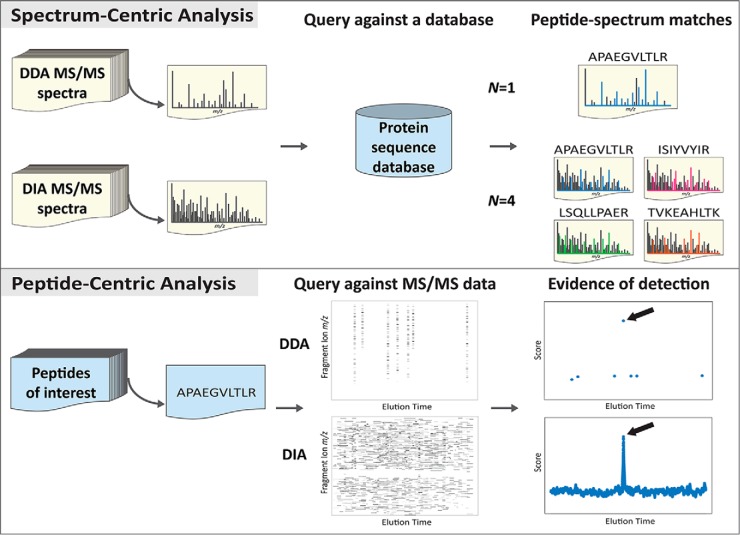
In mass spectrometry-based bottom-up proteomics, data-independent acquisition is an emerging technique because of its comprehensive and unbiased sampling of precursor ions. However, current data-independent acquisition methods use wide precursor isolation windows, resulting in cofragmentation and complex mixture spectra. Thus, conventional database searching tools that identify peptides by interpreting individual tandem MS spectra are inherently limited in analyzing data-independent acquisition data. Here we discuss an alternative approach, peptide-centric analysis, which tests directly for the presence and absence of query peptides. We discuss how peptide-centric analysis resolves some limitations of traditional spectrum-centric analysis, and we outline the unique characteristics of peptide-centric analysis in general.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Gatlin C. L., Eng J. K., Cross S. T., Detter J. C., Yates J. R., 3rd (2000) Automated identification of amino acid sequence variations in proteins by HPLC/microspray tandem mass spectrometry. Anal. Chem. 72, 757–763 - PubMed
-
- Eng J. K., Fischer B., Grossmann J., MacCoss M. J. (2008) A fast SEQUEST cross correlation algorithm. J. Proteome Res. 7, 4598–4602 - PubMed
-
- Koenig T., Menze B. H., Kirchner M., Monigatti F., Parker K. C., Patterson T., Steen J. J., Hamprecht F. A., Steen H. (2008) Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics. J. Proteome Res. 7, 3708–3717 - PubMed
-
- Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 - PubMed
-
- Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
