

Cpipe: a shared variant detection pipeline designed for diagnostic settings
- PMID: 26217397
- PMCID: PMC4515933
- DOI: 10.1186/s13073-015-0191-x
Cpipe: a shared variant detection pipeline designed for diagnostic settings
Abstract
The benefits of implementing high throughput sequencing in the clinic are quickly becoming apparent. However, few freely available bioinformatics pipelines have been built from the ground up with clinical genomics in mind. Here we present Cpipe, a pipeline designed specifically for clinical genetic disease diagnostics. Cpipe was developed by the Melbourne Genomics Health Alliance, an Australian initiative to promote common approaches to genomics across healthcare institutions. As such, Cpipe has been designed to provide fast, effective and reproducible analysis, while also being highly flexible and customisable to meet the individual needs of diverse clinical settings. Cpipe is being shared with the clinical sequencing community as an open source project and is available at http://cpipeline.org.
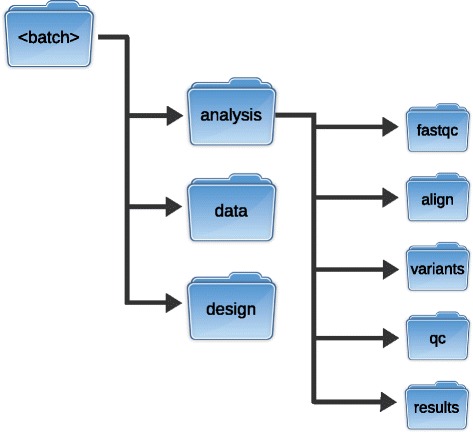
Figures



References
-
- bcbio-nextgen - Validated, scalable, community developed variant calling and RNA-seq analysis. Available at: https://github.com/chapmanb/bcbio-nextgen (accessed 31 March 2015).
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
