

Layered genetic control of DNA methylation and gene expression: a locus of multiple sclerosis in healthy individuals
- PMID: 26220975
- PMCID: PMC4581603
- DOI: 10.1093/hmg/ddv294
Layered genetic control of DNA methylation and gene expression: a locus of multiple sclerosis in healthy individuals
Abstract
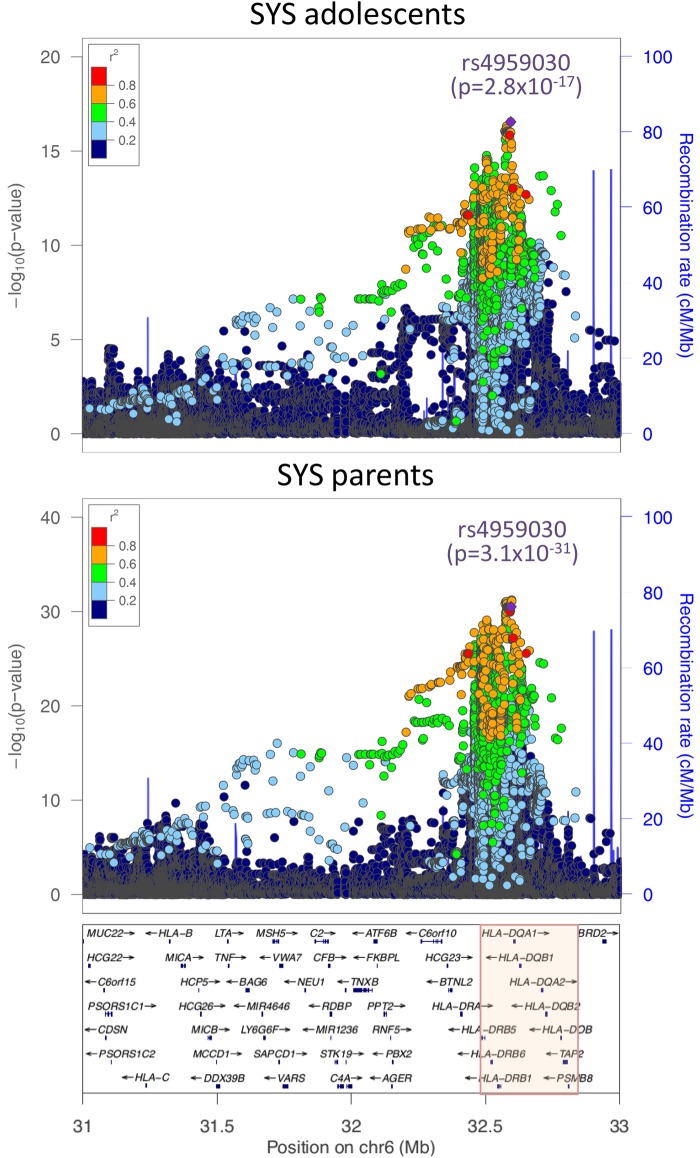
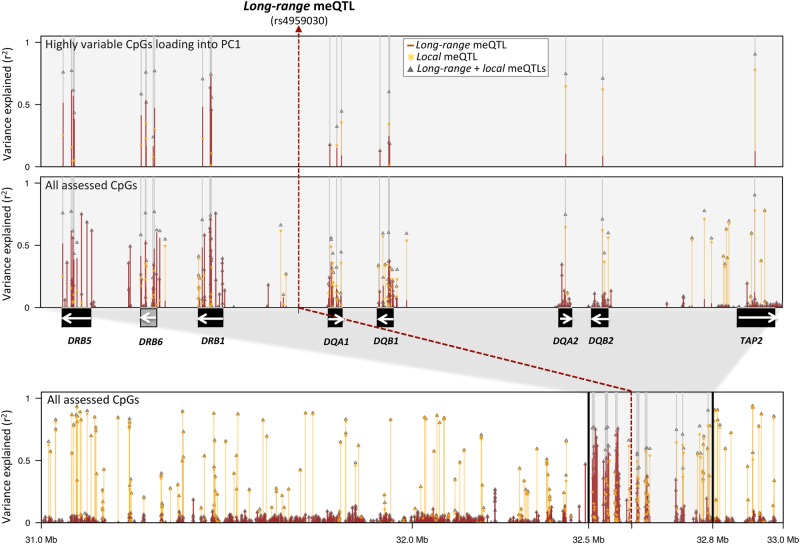
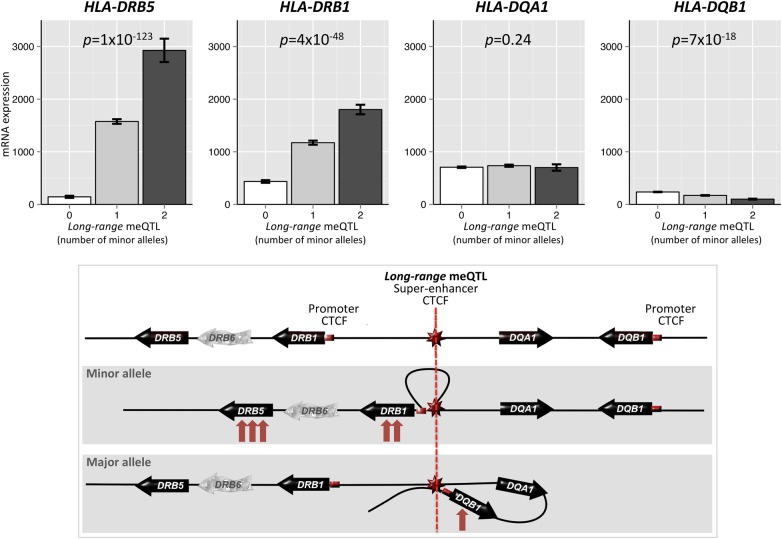
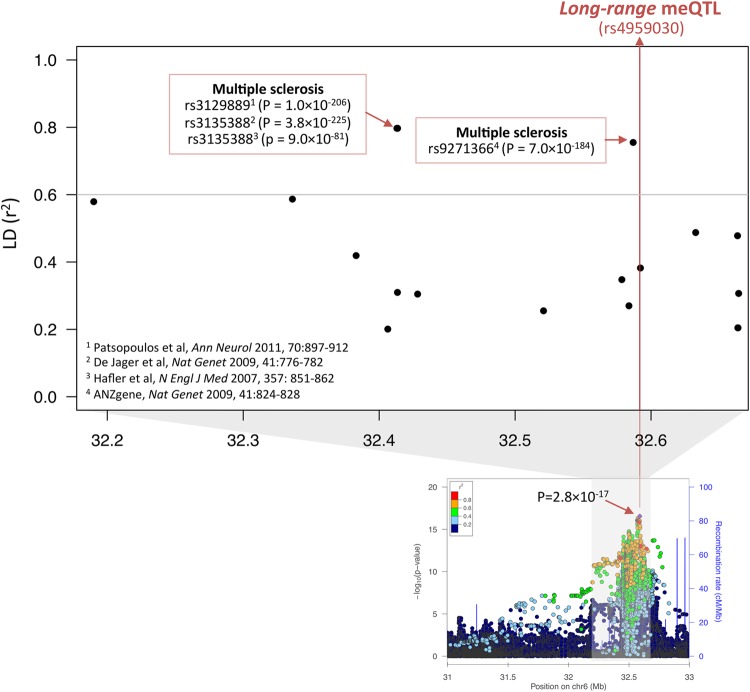
DNA methylation may contribute to the etiology of complex genetic disorders through its impact on genome integrity and gene expression; it is modulated by DNA-sequence variants, named methylation quantitative trait loci (meQTLs). Most meQTLs influence methylation of a few CpG dinucleotides within short genomic regions (<3 kb). Here, we identified a layered genetic control of DNA methylation at numerous CpGs across a long 300 kb genomic region. This control involved a single long-range meQTL and multiple local meQTLs. The long-range meQTL explained up to 75% of variance in methylation of CpGs located over extended areas of the 300 kb region. The meQTL was identified in four samples (P = 2.8 × 10(-17), 3.1 × 10(-31), 4.0 × 10(-71) and 5.2 × 10(-199)), comprising a total of 2796 individuals. The long-range meQTL was strongly associated not only with DNA methylation but also with mRNA expression of several genes within the 300 kb region (P = 7.1 × 10(-18)-1.0 × 10(-123)). The associations of the meQTL with gene expression became attenuated when adjusted for DNA methylation (causal inference test: P = 2.4 × 10(-13)-7.1 × 10(-20)), indicating coordinated regulation of DNA methylation and gene expression. Further, the long-range meQTL was found to be in linkage disequilibrium with the most replicated locus of multiple sclerosis, a disease affecting primarily the brain white matter. In middle-aged adults free of the disease, we observed that the risk allele was associated with subtle structural properties of the brain white matter found in multiple sclerosis (P = 0.02). In summary, we identified a long-range meQTL that controls methylation and expression of several genes and may be involved in increasing brain vulnerability to multiple sclerosis.
© The Author 2015. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures

Similar articles
-
Characterization of cross-tissue genetic-epigenetic effects and their patterns in schizophrenia.Genome Med. 2018 Feb 26;10(1):13. doi: 10.1186/s13073-018-0519-4. Genome Med. 2018. PMID: 29482655 Free PMC article.
-
High density methylation QTL analysis in human blood via next-generation sequencing of the methylated genomic DNA fraction.Genome Biol. 2015 Dec 23;16:291. doi: 10.1186/s13059-015-0842-7. Genome Biol. 2015. PMID: 26699738 Free PMC article.
-
Pancan-meQTL: a database to systematically evaluate the effects of genetic variants on methylation in human cancer.Nucleic Acids Res. 2019 Jan 8;47(D1):D1066-D1072. doi: 10.1093/nar/gky814. Nucleic Acids Res. 2019. PMID: 30203047 Free PMC article.
-
Genetic impacts on DNA methylation: research findings and future perspectives.Genome Biol. 2021 Apr 30;22(1):127. doi: 10.1186/s13059-021-02347-6. Genome Biol. 2021. PMID: 33931130 Free PMC article. Review.
-
From genetic associations to functional studies in multiple sclerosis.Eur J Neurol. 2016 May;23(5):847-53. doi: 10.1111/ene.12981. Epub 2016 Mar 7. Eur J Neurol. 2016. PMID: 26948534 Review.
Cited by
-
Emerging Role for Methylation in Multiple Sclerosis: Beyond DNA.Trends Mol Med. 2017 Jun;23(6):546-562. doi: 10.1016/j.molmed.2017.04.004. Epub 2017 May 4. Trends Mol Med. 2017. PMID: 28478950 Free PMC article. Review.
-
The Emerging Role of Epigenetics in Autoimmune Thyroid Diseases.Front Immunol. 2017 Apr 7;8:396. doi: 10.3389/fimmu.2017.00396. eCollection 2017. Front Immunol. 2017. PMID: 28439272 Free PMC article. Review.
-
DNA methylation as a mediator of HLA-DRB1*15:01 and a protective variant in multiple sclerosis.Nat Commun. 2018 Jun 19;9(1):2397. doi: 10.1038/s41467-018-04732-5. Nat Commun. 2018. PMID: 29921915 Free PMC article.
-
Epigenomics and transcriptomics of systemic sclerosis CD4+ T cells reveal long-range dysregulation of key inflammatory pathways mediated by disease-associated susceptibility loci.Genome Med. 2020 Sep 25;12(1):81. doi: 10.1186/s13073-020-00779-6. Genome Med. 2020. PMID: 32977850 Free PMC article.
-
DNA Methylation: a New Player in Multiple Sclerosis.Mol Neurobiol. 2017 Aug;54(6):4049-4059. doi: 10.1007/s12035-016-9966-3. Epub 2016 Jun 17. Mol Neurobiol. 2017. PMID: 27314687 Review.
References
-
- Kaminsky Z.A., Tang T., Wang S.C., Ptak C., Oh G.H., Wong A.H., Feldcamp L.A., Virtanen C., Halfvarson J., Tysk C. et al. (2009) DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet., 41, 240–245. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
