

A Year of Infection in the Intensive Care Unit: Prospective Whole Genome Sequencing of Bacterial Clinical Isolates Reveals Cryptic Transmissions and Novel Microbiota
- PMID: 26230489
- PMCID: PMC4521703
- DOI: 10.1371/journal.pgen.1005413
A Year of Infection in the Intensive Care Unit: Prospective Whole Genome Sequencing of Bacterial Clinical Isolates Reveals Cryptic Transmissions and Novel Microbiota
Erratum in
-
Correction: A Year of Infection in the Intensive Care Unit: Prospective Whole Genome Sequencing of Bacterial Clinical Isolates Reveals Cryptic Transmissions and Novel Microbiota.PLoS Genet. 2017 Apr 14;13(4):e1006724. doi: 10.1371/journal.pgen.1006724. eCollection 2017 Apr. PLoS Genet. 2017. PMID: 28410416 Free PMC article.
Abstract
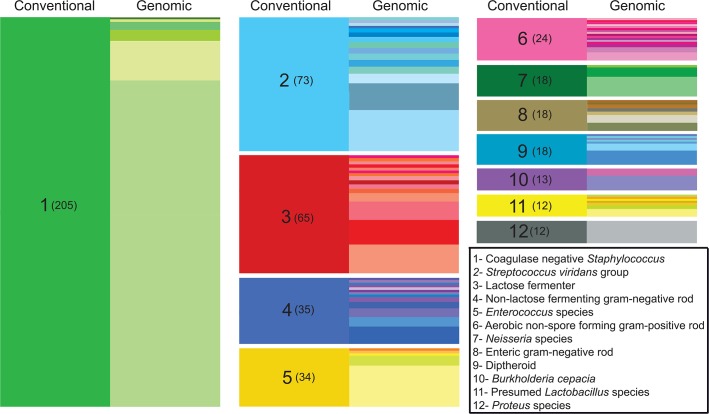
Bacterial whole genome sequencing holds promise as a disruptive technology in clinical microbiology, but it has not yet been applied systematically or comprehensively within a clinical context. Here, over the course of one year, we performed prospective collection and whole genome sequencing of nearly all bacterial isolates obtained from a tertiary care hospital's intensive care units (ICUs). This unbiased collection of 1,229 bacterial genomes from 391 patients enables detailed exploration of several features of clinical pathogens. A sizable fraction of isolates identified as clinically relevant corresponded to previously undescribed species: 12% of isolates assigned a species-level classification by conventional methods actually qualified as distinct, novel genomospecies on the basis of genomic similarity. Pan-genome analysis of the most frequently encountered pathogens in the collection revealed substantial variation in pan-genome size (1,420 to 20,432 genes) and the rate of gene discovery (1 to 152 genes per isolate sequenced). Surprisingly, although potential nosocomial transmission of actively surveilled pathogens was rare, 8.7% of isolates belonged to genomically related clonal lineages that were present among multiple patients, usually with overlapping hospital admissions, and were associated with clinically significant infection in 62% of patients from which they were recovered. Multi-patient clonal lineages were particularly evident in the neonatal care unit, where seven separate Staphylococcus epidermidis clonal lineages were identified, including one lineage associated with bacteremia in 5/9 neonates. Our study highlights key differences in the information made available by conventional microbiological practices versus whole genome sequencing, and motivates the further integration of microbial genome sequencing into routine clinical care.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



References
-
- Council NI (2000) National Intelligence Estimate 99-17D: The Global Infectious Disease Threat and Its Implications for the United States. - PubMed
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
