

Genetic Analysis of Arrhythmogenic Diseases in the Era of NGS: The Complexity of Clinical Decision-Making in Brugada Syndrome
- PMID: 26230511
- PMCID: PMC4521779
- DOI: 10.1371/journal.pone.0133037
Genetic Analysis of Arrhythmogenic Diseases in the Era of NGS: The Complexity of Clinical Decision-Making in Brugada Syndrome
Abstract
Background: The use of next-generation sequencing enables a rapid analysis of many genes associated with sudden cardiac death in diseases like Brugada Syndrome. Genetic variation is identified and associated with 30-35% of cases of Brugada Syndrome, with nearly 20-25% attributable to variants in SCN5A, meaning many cases remain undiagnosed genetically. To evaluate the role of genetic variants in arrhythmogenic diseases and the utility of next-generation sequencing, we applied this technology to resequence 28 main genes associated with arrhythmogenic disorders.
Materials and methods: A cohort of 45 clinically diagnosed Brugada Syndrome patients classified as SCN5A-negative was analyzed using next generation sequencing. Twenty-eight genes were resequenced: AKAP9, ANK2, CACNA1C, CACNB2, CASQ2, CAV3, DSC2, DSG2, DSP, GPD1L, HCN4, JUP, KCNE1, KCNE2, KCNE3, KCNH2, KCNJ2, KCNJ5, KCNQ1, NOS1AP, PKP2, RYR2, SCN1B, SCN3B, SCN4B, SCN5A, SNTA1, and TMEM43. A total of 85 clinically evaluated relatives were also genetically analyzed to ascertain familial segregation.
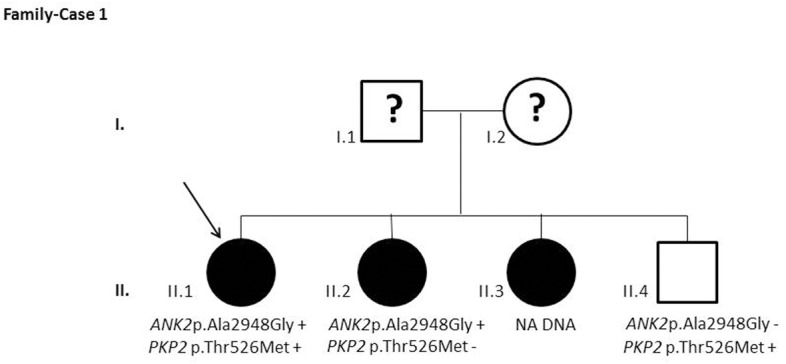
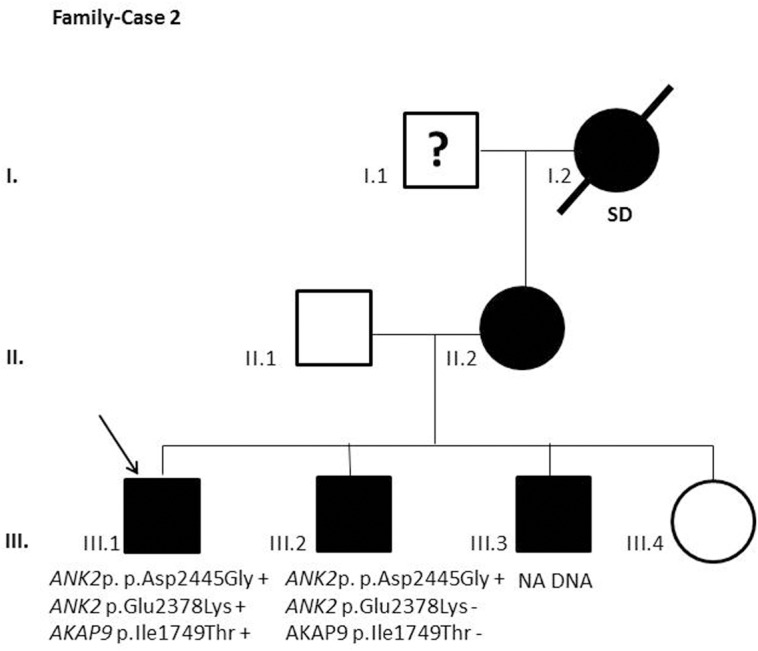
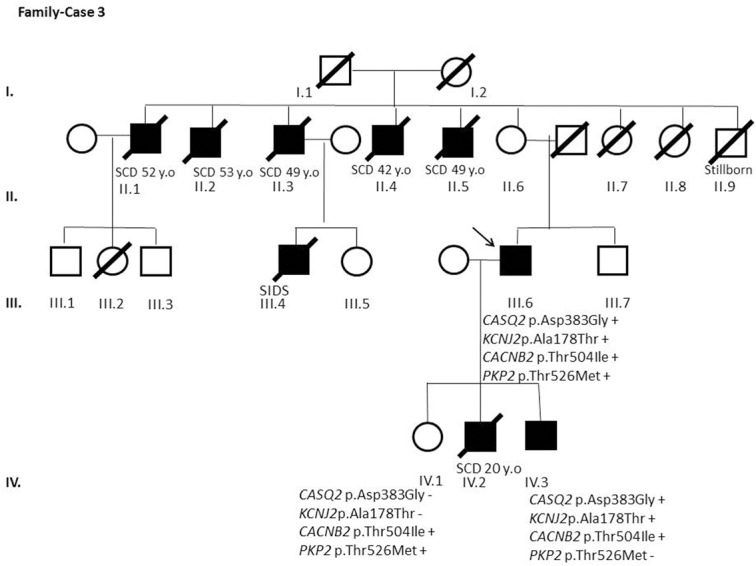
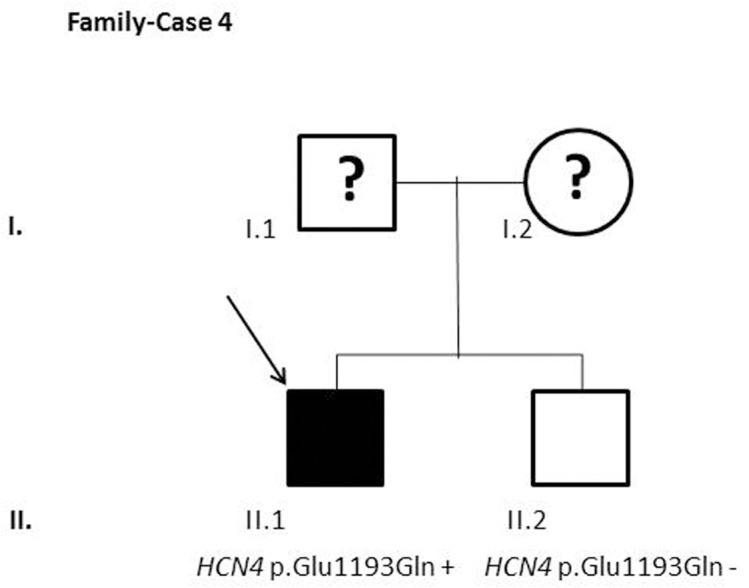
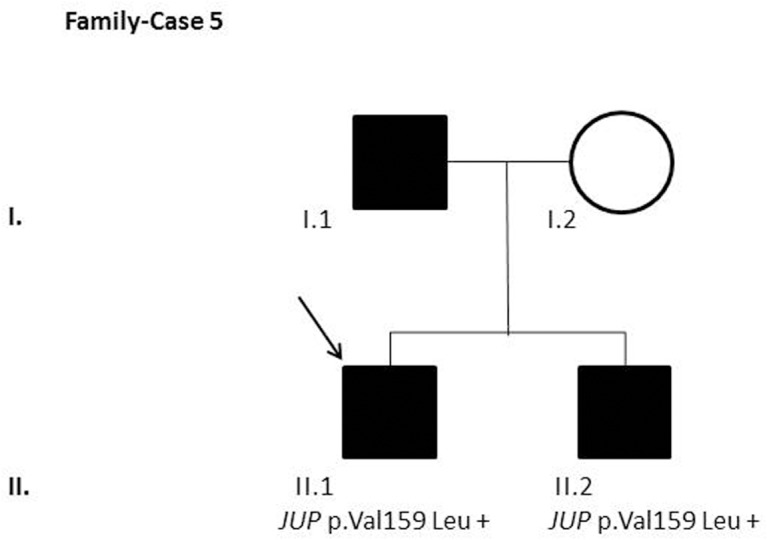
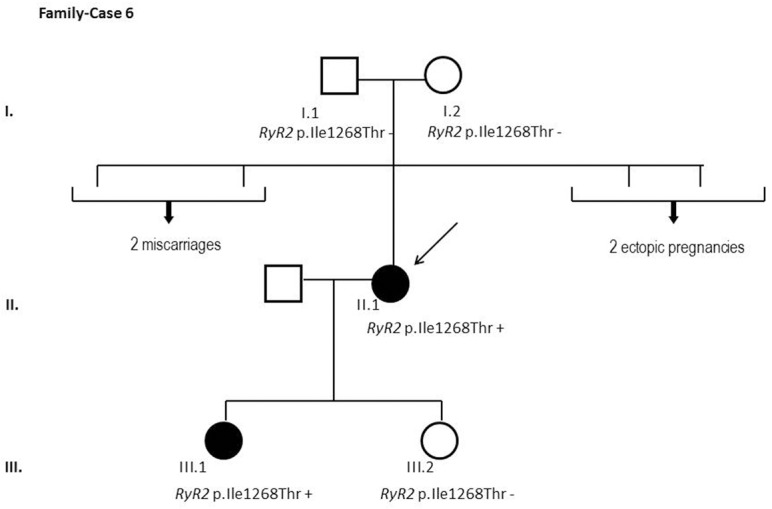
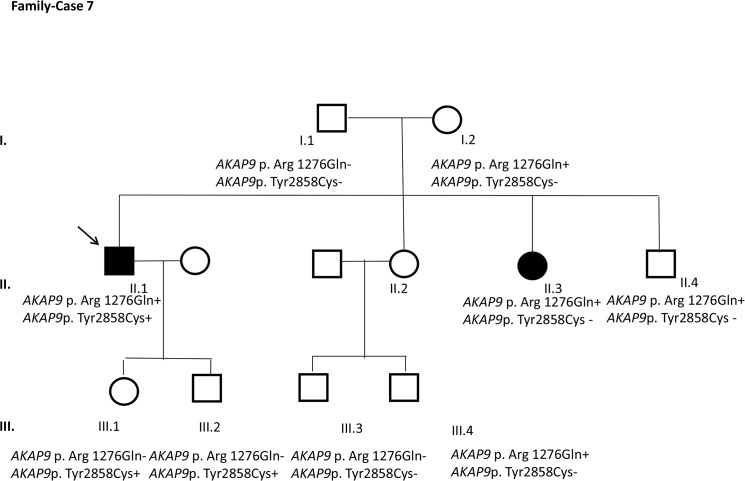
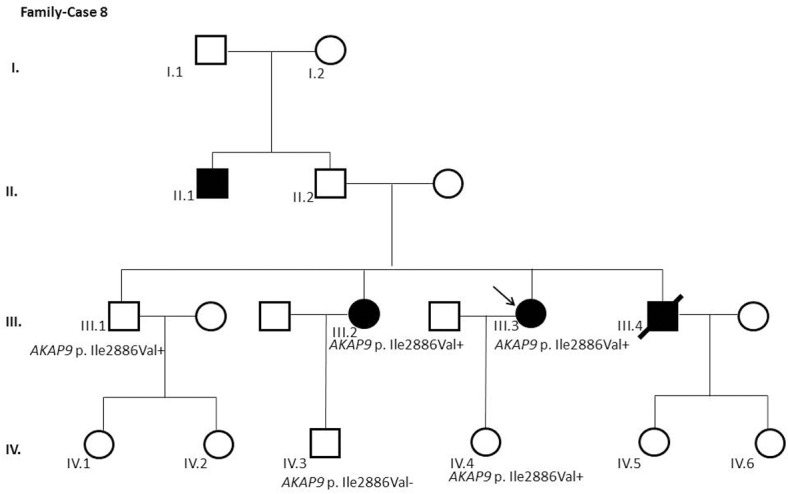
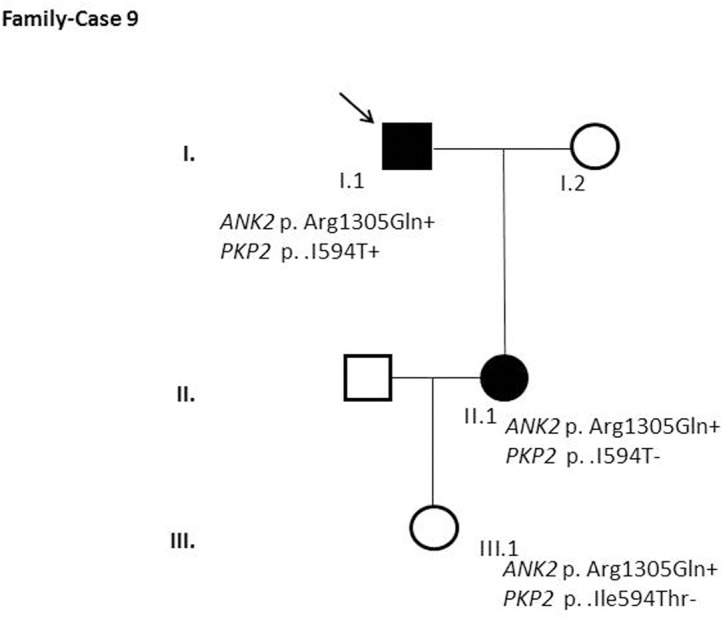
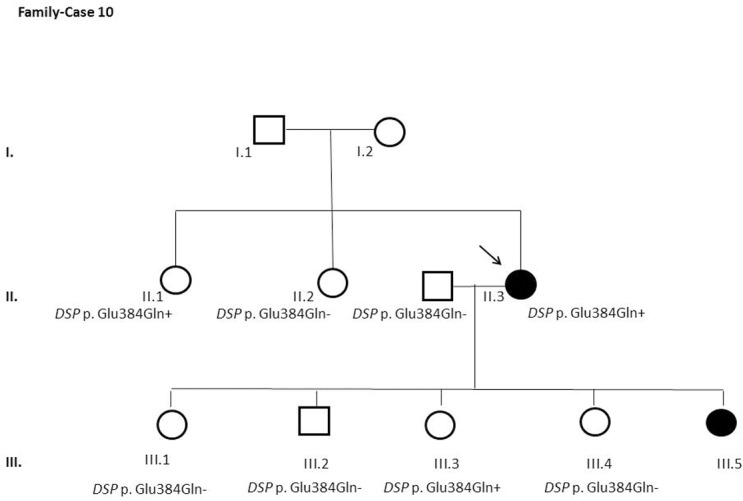
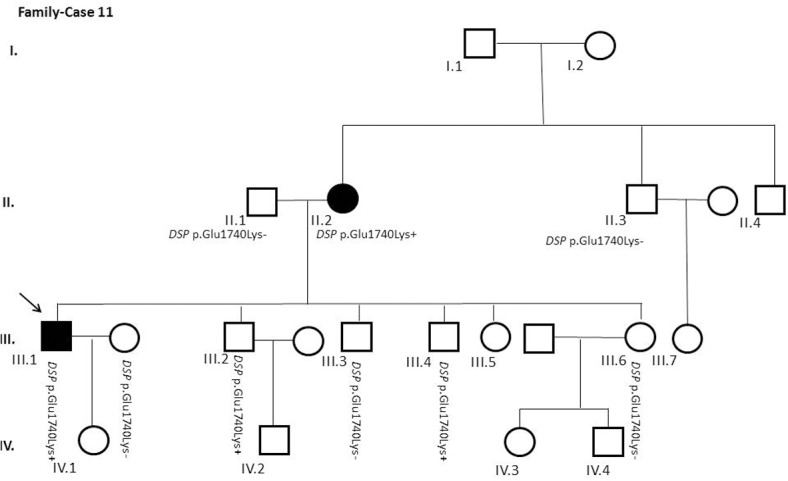
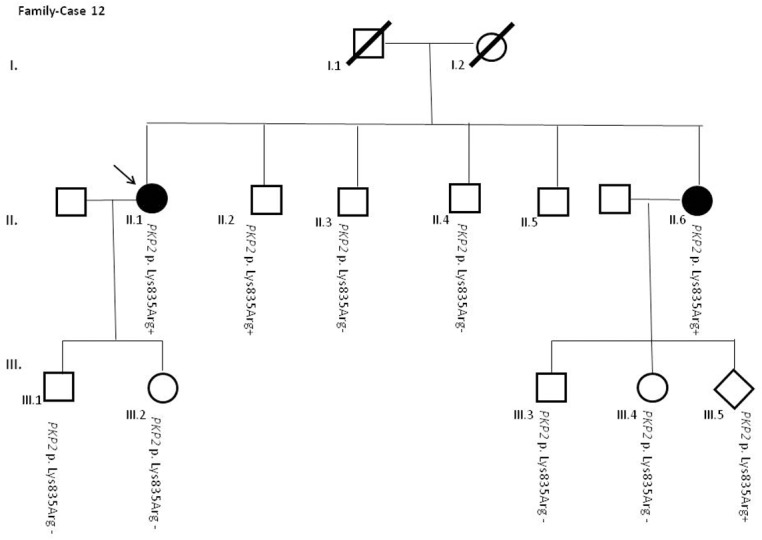
Results and discussion: Twenty-two patients carried 30 rare genetic variants in 12 genes, only 4 of which were previously associated with Brugada Syndrome. Neither insertion/deletion nor copy number variation were detected. We identified genetic variants in novel candidate genes potentially associated to Brugada Syndrome. These include: 4 genetic variations in AKAP9 including a de novo genetic variation in 3 positive cases; 5 genetic variations in ANK2 detected in 4 cases; variations in KCNJ2 together with CASQ2 in 1 case; genetic variations in RYR2, including a de novo genetic variation and desmosomal proteins encoding genes including DSG2, DSP and JUP, detected in 3 of the cases. Larger gene panels or whole exome sequencing should be considered to identify novel genes associated to Brugada Syndrome. However, application of approaches such as whole exome sequencing would difficult the interpretation for clinical purposes due to the large amount of data generated. The identification of these genetic variants opens new perspectives on the implications of genetic background in the arrhythmogenic substrate for research purposes.
Conclusions: As a paradigm for other arrhythmogenic diseases and for unexplained sudden death, our data show that clinical genetic diagnosis is justified in a family perspective for confirmation of genetic causality. In the era of personalized medicine using high-throughput tools, clinical decision-making is increasingly complex.
Conflict of interest statement
Figures
Similar articles
-
Targeted next generation sequencing application in cardiac channelopathies: Analysis of a cohort of autopsy-negative sudden unexplained deaths.Forensic Sci Int. 2015 Sep;254:5-11. doi: 10.1016/j.forsciint.2015.06.023. Epub 2015 Jul 3. Forensic Sci Int. 2015. PMID: 26164358
-
Explaining sudden infant death with cardiac arrhythmias: Complete exon sequencing of nine cardiac arrhythmia genes in Dutch SIDS cases highlights new and known DNA variants.Forensic Sci Int Genet. 2020 May;46:102266. doi: 10.1016/j.fsigen.2020.102266. Epub 2020 Feb 27. Forensic Sci Int Genet. 2020. PMID: 32145446
-
[Genetic and molecular basis for sodium channel-mediated Brugada syndrome].Arch Cardiol Mex. 2013 Oct-Dec;83(4):295-302. doi: 10.1016/j.acmx.2013.10.001. Epub 2013 Nov 21. Arch Cardiol Mex. 2013. PMID: 24269159 Review. Spanish.
-
A balanced translocation disrupting SCN5A in a family with Brugada syndrome and sudden cardiac death.Heart Rhythm. 2019 Feb;16(2):231-238. doi: 10.1016/j.hrthm.2018.08.027. Epub 2018 Aug 28. Heart Rhythm. 2019. PMID: 30170230
-
Update on Genetic Basis of Brugada Syndrome: Monogenic, Polygenic or Oligogenic?Int J Mol Sci. 2020 Sep 28;21(19):7155. doi: 10.3390/ijms21197155. Int J Mol Sci. 2020. PMID: 32998306 Free PMC article. Review.
Cited by
-
The Role of Next-Generation Sequencing in the Management of Patients with Suspected Non-Ischemic Cardiomyopathy after Syncope or Termination of Sudden Arrhythmic Death.Genes (Basel). 2024 Jan 5;15(1):72. doi: 10.3390/genes15010072. Genes (Basel). 2024. PMID: 38254962 Free PMC article.
-
AKAP9-Related Channelopathy: Novel Pathogenic Variant and Review of the Literature.Genes (Basel). 2022 Nov 20;13(11):2167. doi: 10.3390/genes13112167. Genes (Basel). 2022. PMID: 36421840 Free PMC article. Review.
-
Sudden Arrhythmic Death During Exercise: A Post-Mortem Genetic Analysis.Sports Med. 2017 Oct;47(10):2101-2115. doi: 10.1007/s40279-017-0705-3. Sports Med. 2017. PMID: 28255936
-
"Re-evaluation of variants of uncertain significance in patients with hereditary arrhythmogenic disorders".BMC Cardiovasc Disord. 2024 Jul 27;24(1):390. doi: 10.1186/s12872-024-04065-w. BMC Cardiovasc Disord. 2024. PMID: 39068400 Free PMC article.
-
Practical Aspects in Genetic Testing for Cardiomyopathies and Channelopathies.Clin Biochem Rev. 2019 Nov;40(4):187-200. doi: 10.33176/AACB-19-00030. Clin Biochem Rev. 2019. PMID: 31857740 Free PMC article. Review.
References
-
- Brugada R CO, Brugada P. Brugada Syndrome 2005. March 31 [Updated 2012 Aug 16]. In: Pagon RA, Bird TD, Dolan CR, et al. , editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993. Available: http://www.ncbi.nlm.nih.gov/books/NBK1517/. - PubMed
-
- Gollob MH, Blier L, Brugada R, Champagne J, Chauhan V, Connors S, et al. Recommendations for the use of genetic testing in the clinical evaluation of inherited cardiac arrhythmias associated with sudden cardiac death: Canadian Cardiovascular Society/Canadian Heart Rhythm Society joint position paper. The Canadian journal of cardiology. 2011;27(2):232–45. Epub 2011/04/05. 10.1016/j.cjca.2010.12.078 . - DOI - PubMed
-
- Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart rhythm: the official journal of the Heart Rhythm Society. 2013;10(12):1932–63. Epub 2013/09/10. 10.1016/j.hrthm.2013.05.014 . - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
