

The presence of wild type p53 in hematological cancers improves the efficacy of combinational therapy targeting metabolism
- PMID: 26231043
- PMCID: PMC4662487
- DOI: 10.18632/oncotarget.4653
The presence of wild type p53 in hematological cancers improves the efficacy of combinational therapy targeting metabolism
Abstract
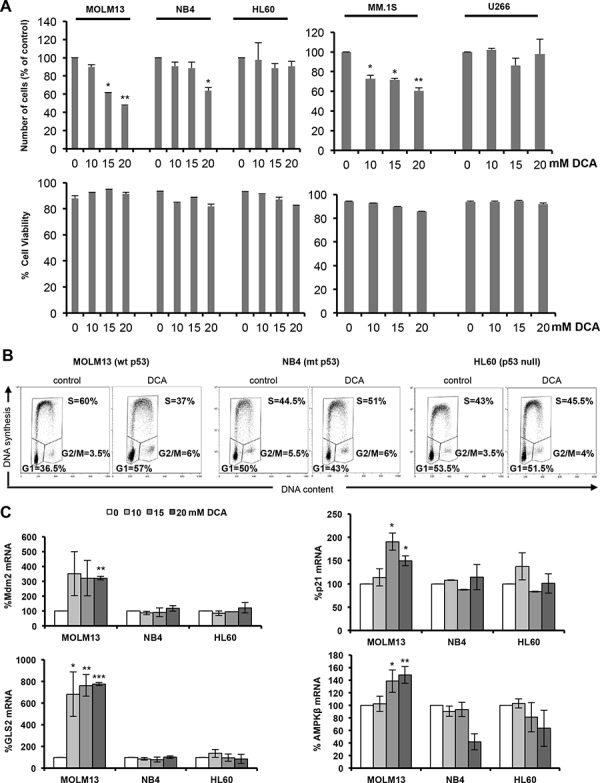
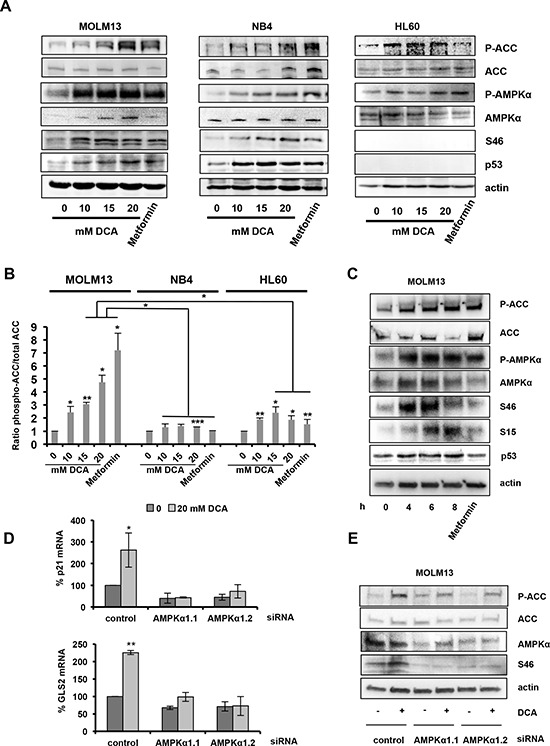
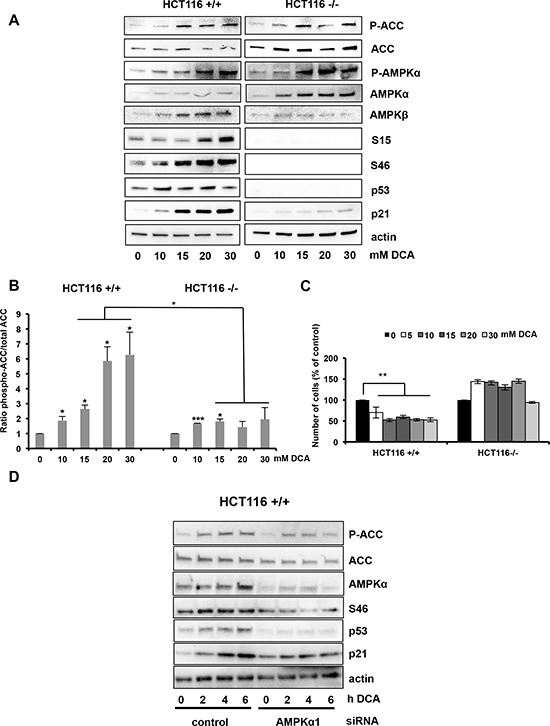
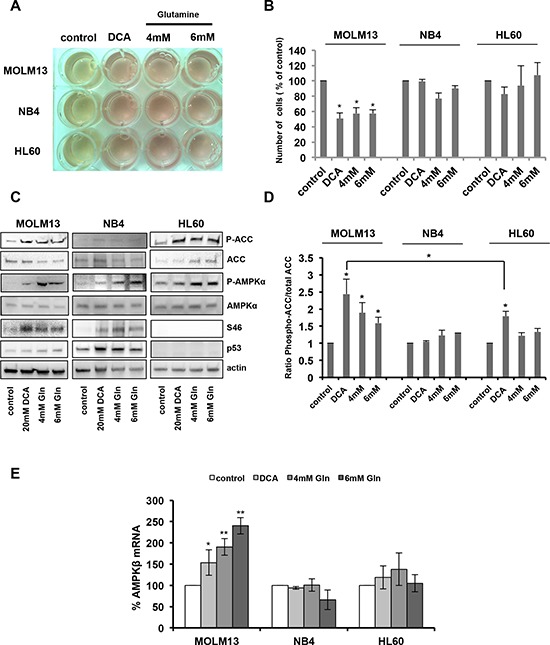
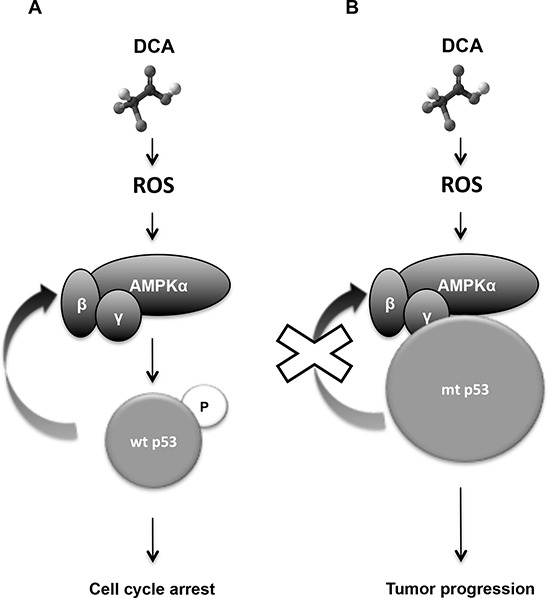
Manipulation of metabolic pathways in hematological cancers has therapeutic potential. Here, we determined the molecular mechanism of action of the metabolic modulator dichloroacetate (DCA) in leukemic cells. We found that DCA induces the AMP-activated protein kinase (AMPK)/p53 pathway with increased efficacy in tumors expressing wild type (wt p53). Clinically relevant, low concentrations of doxorubicin synergize in vitro and in vivo with DCA to further enhance p53 activation and to block tumor progression. Leukemia cell lines and primary leukemic cells containing mutant p53 are resistant to the above-described combination approach. However, DCA synergized with the Hsp90 inhibitor 17-AAG to specifically eliminate these cells. Our studies strongly indicate that depending on the p53 status, different combination therapies would provide better treatment with decreased side effects in hematological cancers.
Keywords: AMPK; dichloroacetate; metabolism; mutant p53; oxidative phosphorylation.
Conflict of interest statement
The authors have filed one patent application EP12305583.2, but declare that they do not have other competing financial or other interests.
Figures
References
-
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. - PubMed
-
- Villalba M, Rathore MG, Lopez-Royuela N, Krzywinska E, Garaude J, Allende-Vega N. From tumor cell metabolism to tumor immune escape. Int J Biochem Cell Biol. 2013;45:106–113. - PubMed
-
- Boag JM, Beesley AH, Firth MJ, Freitas JR, Ford J, Hoffmann K, Cummings AJ, de Klerk NH, Kees UR. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leukemia. 2006;20:1731–1737. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
