

Identification of Genetic Factors that Modify Clinical Onset of Huntington's Disease
- PMID: 26232222
- PMCID: PMC4524551
- DOI: 10.1016/j.cell.2015.07.003
Identification of Genetic Factors that Modify Clinical Onset of Huntington's Disease
Abstract
As a Mendelian neurodegenerative disorder, the genetic risk of Huntington's disease (HD) is conferred entirely by an HTT CAG repeat expansion whose length is the primary determinant of the rate of pathogenesis leading to disease onset. To investigate the pathogenic process that precedes disease, we used genome-wide association (GWA) analysis to identify loci harboring genetic variations that alter the age at neurological onset of HD. A chromosome 15 locus displays two independent effects that accelerate or delay onset by 6.1 years and 1.4 years, respectively, whereas a chromosome 8 locus hastens onset by 1.6 years. Association at MLH1 and pathway analysis of the full GWA results support a role for DNA handling and repair mechanisms in altering the course of HD. Our findings demonstrate that HD disease modification in humans occurs in nature and offer a genetic route to identifying in-human validated therapeutic targets in this and other Mendelian disorders.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
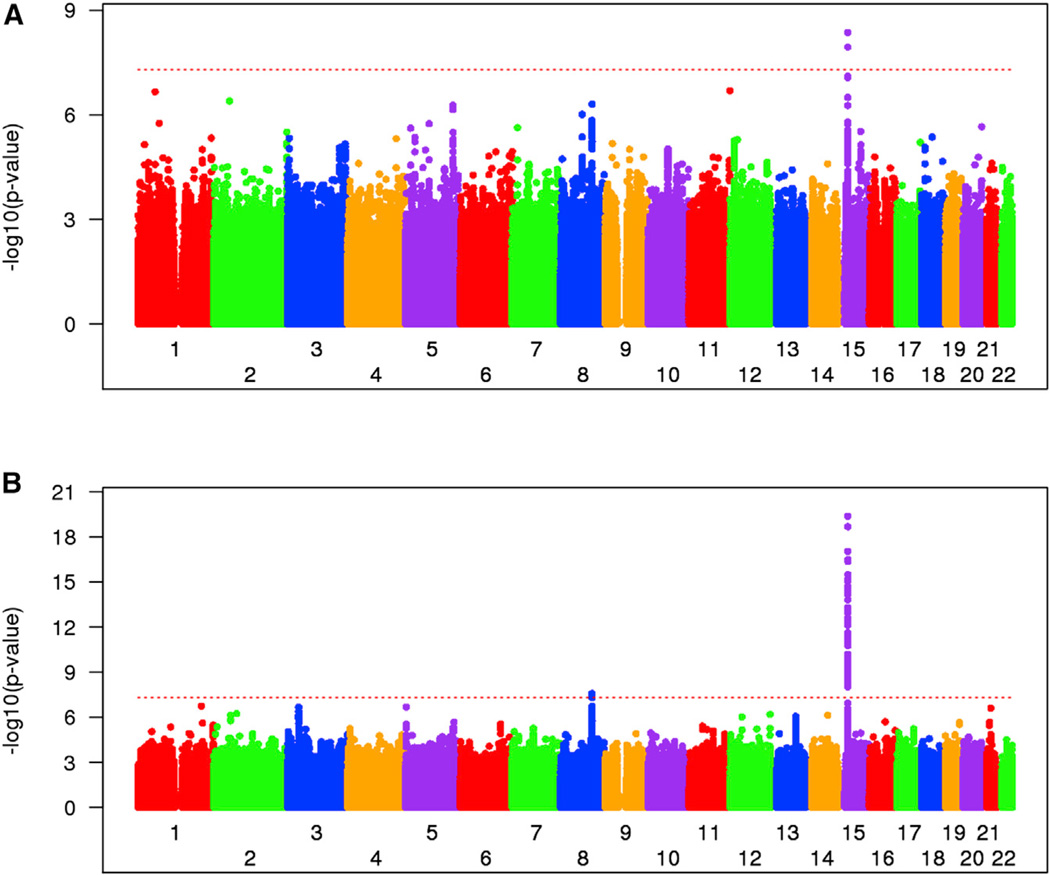
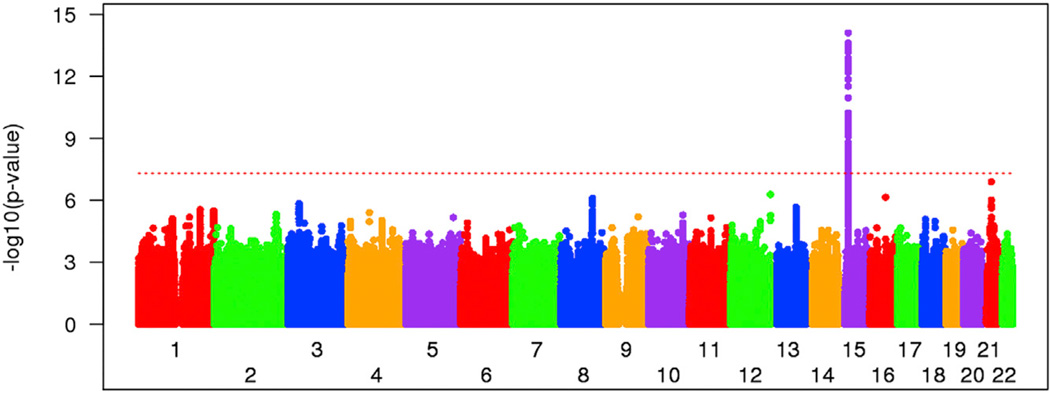
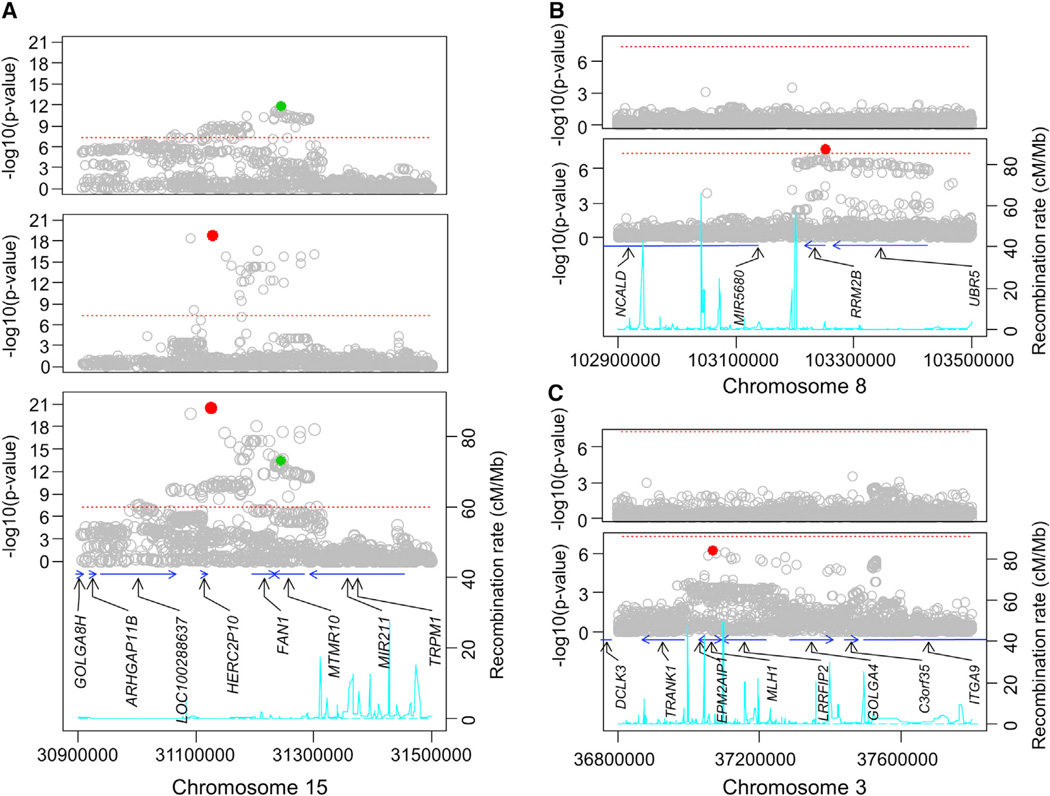
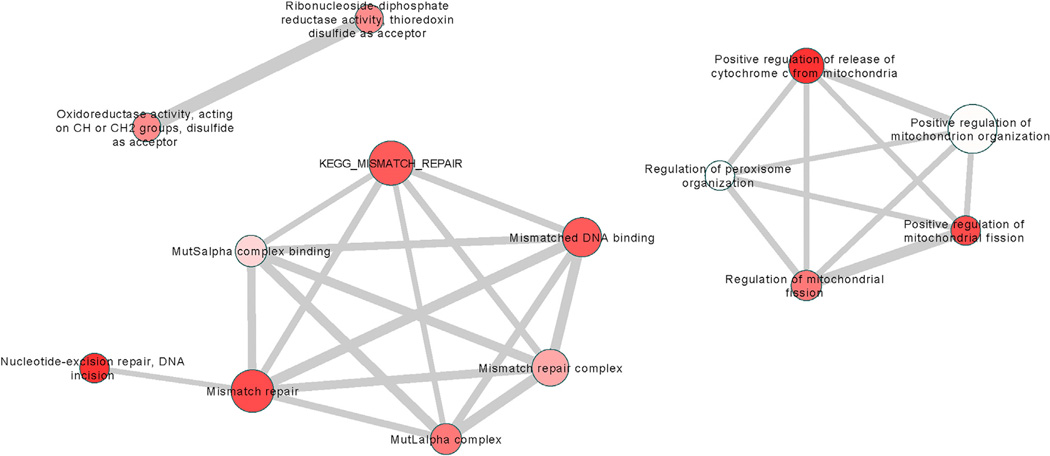
References
-
- Aronin N, DiFiglia M. Huntingtin-lowering strategies in Huntington’s disease: antisense oligonucleotides, small RNAs, and gene editing. Mov. Disord. 2014;29:1455–1461. - PubMed
-
- Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chrétien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007;39:776–780. - PubMed
-
- Brown MB. A method for combining non-independent, one-sided tests of significance. Biometrics. 1975;31:978–992.
Publication types
MeSH terms
Substances
Grants and funding
- R01HG002449/HG/NHGRI NIH HHS/United States
- P50NS016367/NS/NINDS NIH HHS/United States
- U01 NS082079/NS/NINDS NIH HHS/United States
- R01 NS040068/NS/NINDS NIH HHS/United States
- R01 NS091161/NS/NINDS NIH HHS/United States
- P50 NS016367/NS/NINDS NIH HHS/United States
- U01NS082079/NS/NINDS NIH HHS/United States
- G0801418/MRC_/Medical Research Council/United Kingdom
- X01HG006074/HG/NHGRI NIH HHS/United States
- R01 HG002449/HG/NHGRI NIH HHS/United States
- R01NS091161/NS/NINDS NIH HHS/United States
- MR/L012936/1/MRC_/Medical Research Council/United Kingdom
- R01 NS073947/NS/NINDS NIH HHS/United States
- MR/L010305/1/MRC_/Medical Research Council/United Kingdom
- MR/L023784/2/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
