

Connecting free energy surfaces in implicit and explicit solvent: an efficient method to compute conformational and solvation free energies
- PMID: 26236174
- PMCID: PMC4521639
- DOI: 10.1021/acs.jctc.5b00264
Connecting free energy surfaces in implicit and explicit solvent: an efficient method to compute conformational and solvation free energies
Abstract
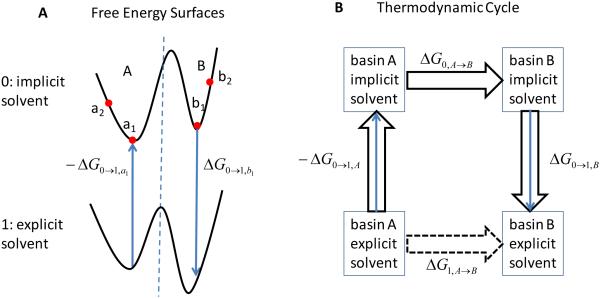
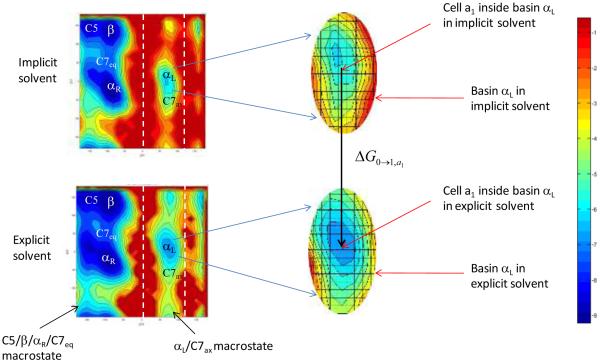
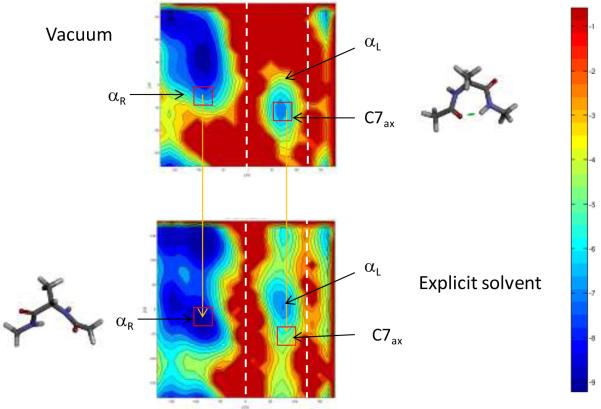
The ability to accurately model solvent effects on free energy surfaces is important for understanding many biophysical processes including protein folding and misfolding, allosteric transitions, and protein–ligand binding. Although all-atom simulations in explicit solvent can provide an accurate model for biomolecules in solution, explicit solvent simulations are hampered by the slow equilibration on rugged landscapes containing multiple basins separated by barriers. In many cases, implicit solvent models can be used to significantly speed up the conformational sampling; however, implicit solvent simulations do not fully capture the effects of a molecular solvent, and this can lead to loss of accuracy in the estimated free energies. Here we introduce a new approach to compute free energy changes in which the molecular details of explicit solvent simulations are retained while also taking advantage of the speed of the implicit solvent simulations. In this approach, the slow equilibration in explicit solvent, due to the long waiting times before barrier crossing, is avoided by using a thermodynamic cycle which connects the free energy basins in implicit solvent and explicit solvent using a localized decoupling scheme. We test this method by computing conformational free energy differences and solvation free energies of the model system alanine dipeptide in water. The free energy changes between basins in explicit solvent calculated using fully explicit solvent paths agree with the corresponding free energy differences obtained using the implicit/explicit thermodynamic cycle to within 0.3 kcal/mol out of ∼3 kcal/mol at only ∼8% of the computational cost. We note that WHAM methods can be used to further improve the efficiency and accuracy of the implicit/explicit thermodynamic cycle.
Figures
Similar articles
-
Advances in implicit models of water solvent to compute conformational free energy and molecular dynamics of proteins at constant pH.Adv Protein Chem Struct Biol. 2011;85:281-322. doi: 10.1016/B978-0-12-386485-7.00008-9. Adv Protein Chem Struct Biol. 2011. PMID: 21920327 Review.
-
Discrimination between native and intentionally misfolded conformations of proteins: ES/IS, a new method for calculating conformational free energy that uses both dynamics simulations with an explicit solvent and an implicit solvent continuum model.Proteins. 1998 Sep 1;32(4):399-413. Proteins. 1998. PMID: 9726412
-
Free energies of solvation in the context of protein folding: Implications for implicit and explicit solvent models.J Comput Chem. 2016 Mar 15;37(7):629-40. doi: 10.1002/jcc.24235. Epub 2015 Nov 12. J Comput Chem. 2016. PMID: 26558440
-
Accurate calculation of conformational free energy differences in explicit water: the confinement-solvation free energy approach.J Phys Chem B. 2015 Apr 23;119(16):5194-207. doi: 10.1021/acs.jpcb.5b01632. Epub 2015 Apr 9. J Phys Chem B. 2015. PMID: 25807150
-
Implicit modeling of nonpolar solvation for simulating protein folding and conformational transitions.Phys Chem Chem Phys. 2008 Jan 28;10(4):471-81. doi: 10.1039/b714141f. Epub 2007 Nov 14. Phys Chem Chem Phys. 2008. PMID: 18183310 Review.
Cited by
-
Spatially-Decomposed Free Energy of Solvation Based on the Endpoint Density-Functional Method.J Chem Theory Comput. 2019 May 14;15(5):2896-2912. doi: 10.1021/acs.jctc.8b01309. Epub 2019 Apr 16. J Chem Theory Comput. 2019. PMID: 30990682 Free PMC article.
-
Improvements to the ABSINTH Force Field for Proteins Based on Experimentally Derived Amino Acid Specific Backbone Conformational Statistics.J Chem Theory Comput. 2019 Feb 12;15(2):1367-1382. doi: 10.1021/acs.jctc.8b00573. Epub 2019 Jan 22. J Chem Theory Comput. 2019. PMID: 30633502 Free PMC article.
-
Computing conformational free energy differences in explicit solvent: An efficient thermodynamic cycle using an auxiliary potential and a free energy functional constructed from the end points.J Comput Chem. 2017 Jun 5;38(15):1198-1208. doi: 10.1002/jcc.24668. Epub 2016 Dec 23. J Comput Chem. 2017. PMID: 28008630 Free PMC article.
-
Stratified UWHAM and Its Stochastic Approximation for Multicanonical Simulations Which Are Far from Equilibrium.J Chem Theory Comput. 2017 Oct 10;13(10):4660-4674. doi: 10.1021/acs.jctc.7b00651. Epub 2017 Sep 28. J Chem Theory Comput. 2017. PMID: 28902500 Free PMC article.
-
Conformational Free Energy Changes via an Alchemical Path without Reaction Coordinates.J Phys Chem Lett. 2018 Aug 2;9(15):4428-4435. doi: 10.1021/acs.jpclett.8b01851. Epub 2018 Jul 24. J Phys Chem Lett. 2018. PMID: 30024165 Free PMC article.
References
-
- Simonson T. computational biochemistry and biophysics. Marcel Dekker; New York: 2000. Free Energy Calculations.
-
- Chipot C, Pohorille A, editors. Springer series in chemical physics. Springer; New York: 2007. Free Energy Calculations: Theory and Applications in Chemistry and Biology, Study.
-
- Hansen N, van Gunsteren WF. Practical Aspects of Free-Energy Calculations: A Review. J. Chem. Theory Comput. 2014;10(7):2632–2647. - PubMed
-
- Hansmann UHE. Parallel Tempering Algorithm for Conformational Studies of Biological Molecules. Chem. Phys. Lett. 1997;281(1-3):140–150.
-
- Sugita Y, Okamoto Y. Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett. 1999;314(1-2):141–151.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources