

Molecular pathophysiology and pharmacology of the voltage-sensing module of neuronal ion channels
- PMID: 26236192
- PMCID: PMC4502356
- DOI: 10.3389/fncel.2015.00259
Molecular pathophysiology and pharmacology of the voltage-sensing module of neuronal ion channels
Abstract
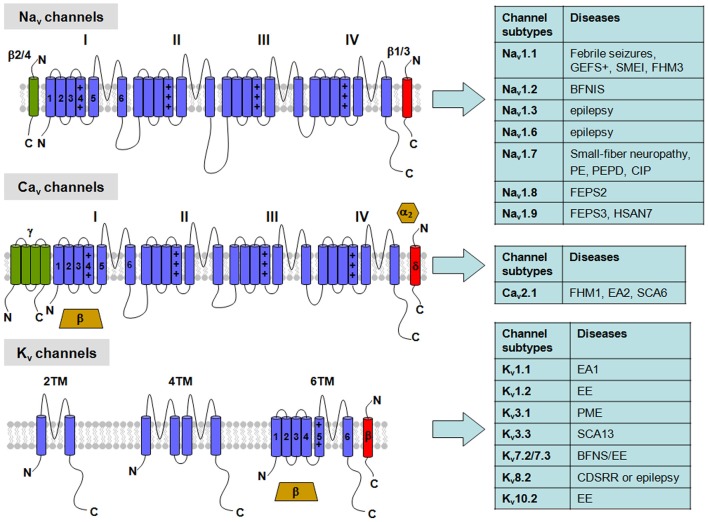
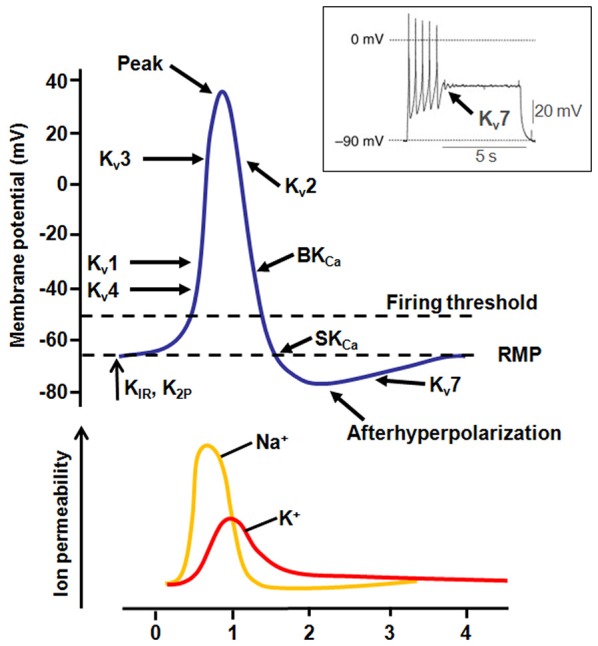
Voltage-gated ion channels (VGICs) are membrane proteins that switch from a closed to open state in response to changes in membrane potential, thus enabling ion fluxes across the cell membranes. The mechanism that regulate the structural rearrangements occurring in VGICs in response to changes in membrane potential still remains one of the most challenging topic of modern biophysics. Na(+), Ca(2+) and K(+) voltage-gated channels are structurally formed by the assembly of four similar domains, each comprising six transmembrane segments. Each domain can be divided into two main regions: the Pore Module (PM) and the Voltage-Sensing Module (VSM). The PM (helices S5 and S6 and intervening linker) is responsible for gate opening and ion selectivity; by contrast, the VSM, comprising the first four transmembrane helices (S1-S4), undergoes the first conformational changes in response to membrane voltage variations. In particular, the S4 segment of each domain, which contains several positively charged residues interspersed with hydrophobic amino acids, is located within the membrane electric field and plays an essential role in voltage sensing. In neurons, specific gating properties of each channel subtype underlie a variety of biological events, ranging from the generation and propagation of electrical impulses, to the secretion of neurotransmitters and to the regulation of gene expression. Given the important functional role played by the VSM in neuronal VGICs, it is not surprising that various VSM mutations affecting the gating process of these channels are responsible for human diseases, and that compounds acting on the VSM have emerged as important investigational tools with great therapeutic potential. In the present review we will briefly describe the most recent discoveries concerning how the VSM exerts its function, how genetically inherited diseases caused by mutations occurring in the VSM affects gating in VGICs, and how several classes of drugs and toxins selectively target the VSM.
Keywords: channelopathies; gating modifier; ion channels; mutations; voltage-sensing module.
Figures

Similar articles
-
Emerging issues of connexin channels: biophysics fills the gap.Q Rev Biophys. 2001 Aug;34(3):325-472. doi: 10.1017/s0033583501003705. Q Rev Biophys. 2001. PMID: 11838236 Review.
-
Voltage-dependent gating of hyperpolarization-activated, cyclic nucleotide-gated pacemaker channels: molecular coupling between the S4-S5 and C-linkers.J Biol Chem. 2004 Apr 2;279(14):13859-65. doi: 10.1074/jbc.M313704200. Epub 2004 Jan 15. J Biol Chem. 2004. PMID: 14726518
-
A complicated complex: Ion channels, voltage sensing, cell membranes and peptide inhibitors.Neurosci Lett. 2018 Jul 13;679:35-47. doi: 10.1016/j.neulet.2018.04.030. Epub 2018 Apr 21. Neurosci Lett. 2018. PMID: 29684532 Review.
-
Opposite Effects of the S4-S5 Linker and PIP(2) on Voltage-Gated Channel Function: KCNQ1/KCNE1 and Other Channels.Front Pharmacol. 2012 Jul 5;3:125. doi: 10.3389/fphar.2012.00125. eCollection 2012. Front Pharmacol. 2012. PMID: 22787448 Free PMC article.
-
Role of hydrophobic and ionic forces in the movement of S4 of the Shaker potassium channel.Mol Membr Biol. 2012 Dec;29(8):321-32. doi: 10.3109/09687688.2012.710343. Epub 2012 Aug 13. Mol Membr Biol. 2012. PMID: 22881396
Cited by
-
Ion channel long non-coding RNAs in neuropathic pain.Pflugers Arch. 2022 Apr;474(4):457-468. doi: 10.1007/s00424-022-02675-x. Epub 2022 Mar 2. Pflugers Arch. 2022. PMID: 35235008 Review.
-
Effect of Short-Term Restraint Stress on the Hypothalamic Transcriptome Profiles of Rats with Inherited Stress-Induced Arterial Hypertension (ISIAH) and Normotensive Wistar Albino Glaxo (WAG) Rats.Int J Mol Sci. 2024 Jun 18;25(12):6680. doi: 10.3390/ijms25126680. Int J Mol Sci. 2024. PMID: 38928385 Free PMC article.
-
Clinical Spectrum of KCNA1 Mutations: New Insights into Episodic Ataxia and Epilepsy Comorbidity.Int J Mol Sci. 2020 Apr 17;21(8):2802. doi: 10.3390/ijms21082802. Int J Mol Sci. 2020. PMID: 32316562 Free PMC article. Review.
-
De novo KCNH1 mutations in four patients with syndromic developmental delay, hypotonia and seizures.J Hum Genet. 2016 May;61(5):381-7. doi: 10.1038/jhg.2016.1. Epub 2016 Jan 28. J Hum Genet. 2016. PMID: 26818738 Review.
-
Therapeutic Approaches to Genetic Ion Channelopathies and Perspectives in Drug Discovery.Front Pharmacol. 2016 May 10;7:121. doi: 10.3389/fphar.2016.00121. eCollection 2016. Front Pharmacol. 2016. PMID: 27242528 Free PMC article. Review.
References
-
- Baasch A. L., Hüning I., Gilissen C., Klepper J., Veltman J. A., Gillessen-Kaesbach G., et al. . (2014). Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia and brain abnormalities. Epilepsia 55, e25–29. 10.1111/epi.12554 - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous