

ViennaNGS: A toolbox for building efficient next- generation sequencing analysis pipelines
- PMID: 26236465
- PMCID: PMC4513691
- DOI: 10.12688/f1000research.6157.2
ViennaNGS: A toolbox for building efficient next- generation sequencing analysis pipelines
Abstract
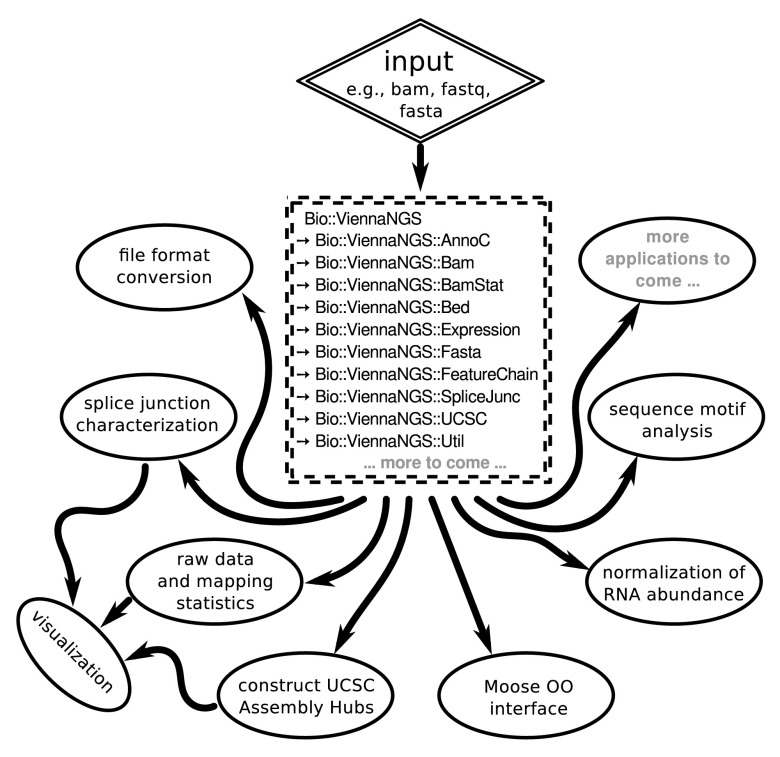
Recent achievements in next-generation sequencing (NGS) technologies lead to a high demand for reuseable software components to easily compile customized analysis workflows for big genomics data. We present ViennaNGS, an integrated collection of Perl modules focused on building efficient pipelines for NGS data processing. It comes with functionality for extracting and converting features from common NGS file formats, computation and evaluation of read mapping statistics, as well as normalization of RNA abundance. Moreover, ViennaNGS provides software components for identification and characterization of splice junctions from RNA-seq data, parsing and condensing sequence motif data, automated construction of Assembly and Track Hubs for the UCSC genome browser, as well as wrapper routines for a set of commonly used NGS command line tools.
Keywords: Perl; RNA-seq; next generation sequencing; pipelines; read mapping.
Conflict of interest statement
Figures


References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases