

Congenital generalized lipodystrophies--new insights into metabolic dysfunction
- PMID: 26239609
- PMCID: PMC7605893
- DOI: 10.1038/nrendo.2015.123
Congenital generalized lipodystrophies--new insights into metabolic dysfunction
Abstract
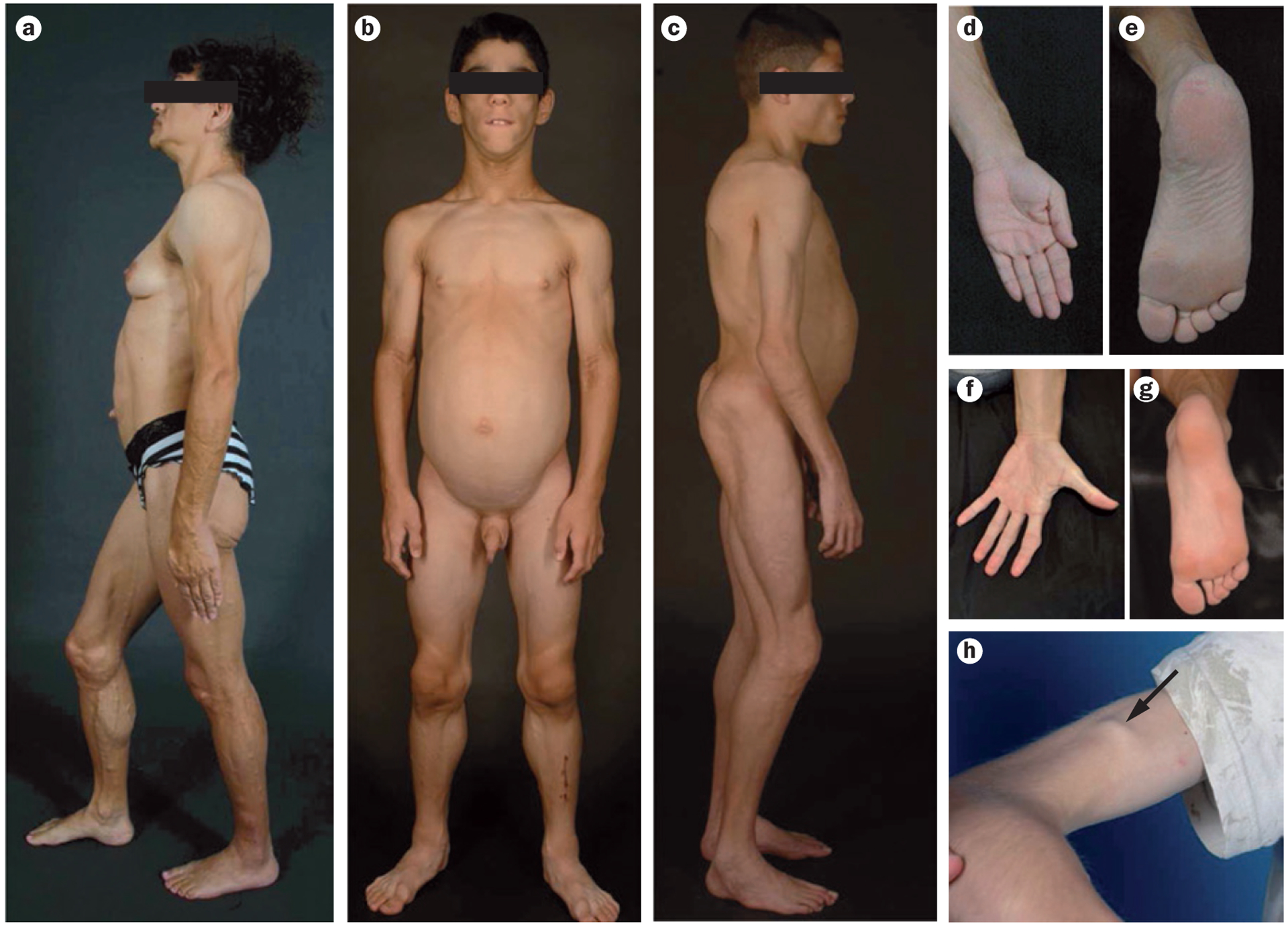
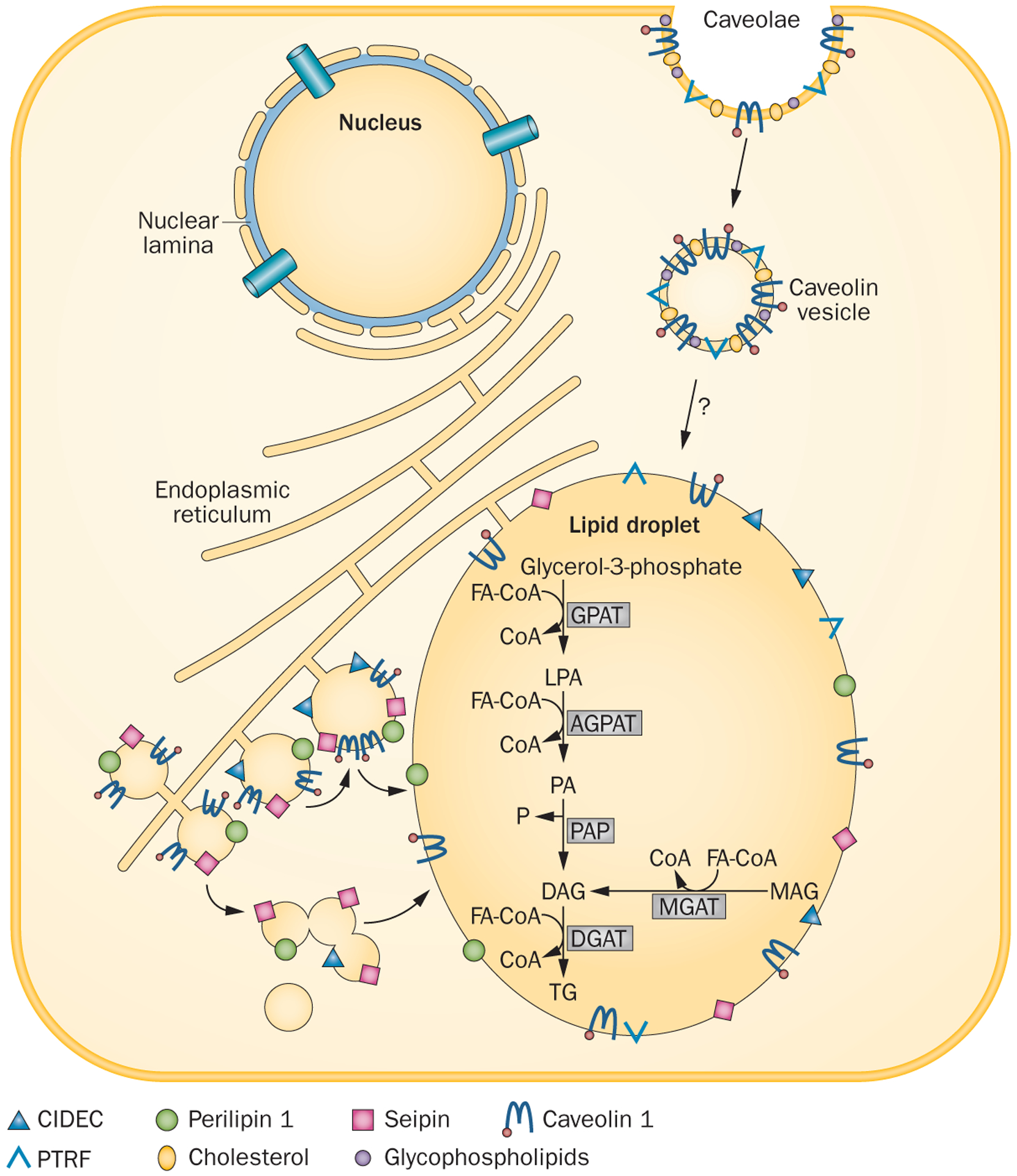
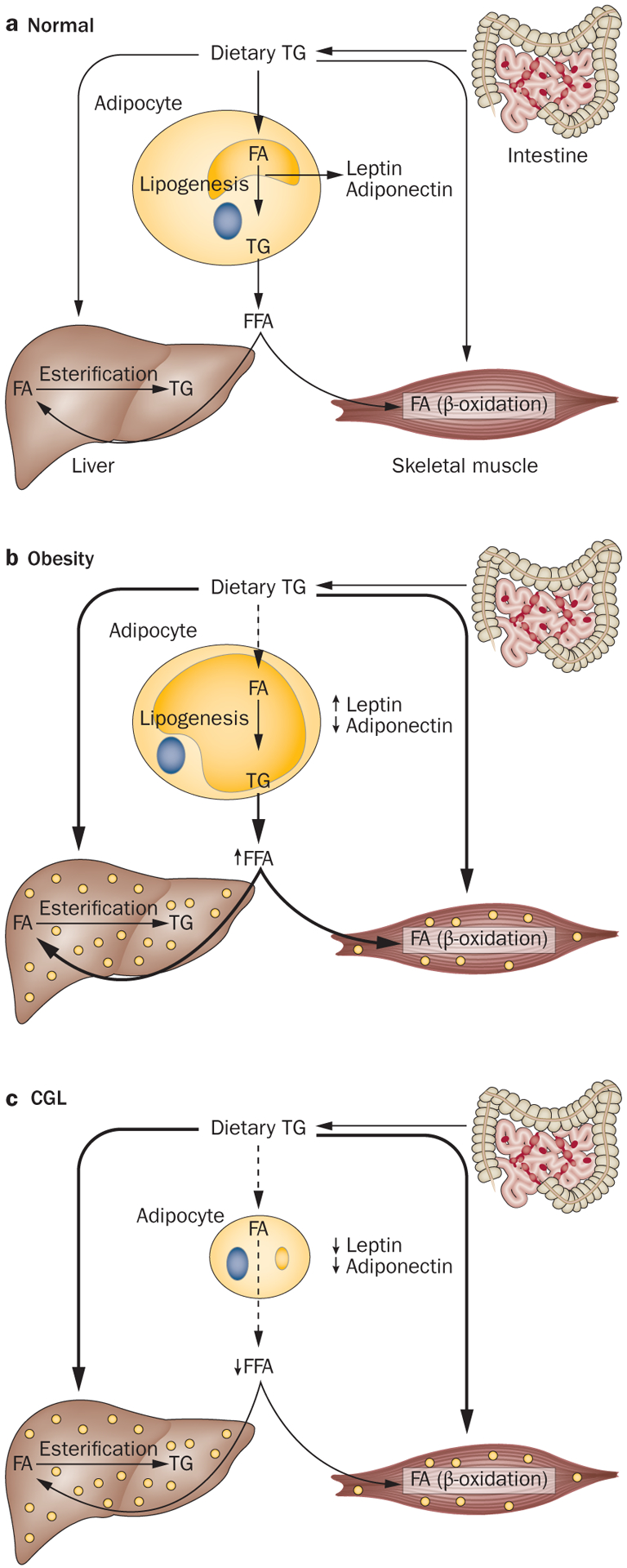
Congenital generalized lipodystrophy (CGL) is a heterogeneous autosomal recessive disorder characterized by a near complete lack of adipose tissue from birth and, later in life, the development of metabolic complications, such as diabetes mellitus, hypertriglyceridaemia and hepatic steatosis. Four distinct subtypes of CGL exist: type 1 is associated with AGPAT2 mutations; type 2 is associated with BSCL2 mutations; type 3 is associated with CAV1 mutations; and type 4 is associated with PTRF mutations. The products of these genes have crucial roles in phospholipid and triglyceride synthesis, as well as in the formation of lipid droplets and caveolae within adipocytes. The predominant cause of metabolic complications in CGL is excess triglyceride accumulation in the liver and skeletal muscle owing to the inability to store triglycerides in adipose tissue. Profound hypoleptinaemia further exacerbates metabolic derangements by inducing a voracious appetite. Patients require psychological support, a low-fat diet, increased physical activity and cosmetic surgery. Aside from conventional therapy for hyperlipidaemia and diabetes mellitus, metreleptin replacement therapy can dramatically improve metabolic complications in patients with CGL. In this Review, we discuss the molecular genetic basis of CGL, the pathogenesis of the disease's metabolic complications and therapeutic options for patients with CGL.
Conflict of interest statement
Competing interests
A.G. co-holds a patent regarding the use of leptin for treating human lipoatrophy and the method of determining predisposition to this treatment but receives no financial compensation. A.G. has received research grants from Aegerion, Astra-Zeneca, Bristol-Myers-Squibb and Pfizer and is a consultant for Amgen, Back Bay Life Sciences, Biomarin Pharmaceuticals, BioMedical Insights, Clearview Healthcare, Eli Lilly, Engage Health, Gerson Lehrman Group, Health Advances, Ipsen Pharmaceuticals, Intellisphere, Medscape and Tekmira. A.G. is also an advisory board member for AstraZeneca. N.P. declares no competing interests.
Figures
References
-
- Garg A Acquired and inherited lipodystrophies. N. Engl. J. Med 350, 1220–1234 (2004). - PubMed
-
- Berardinelli W An undiagnosed endocrinometabolic syndrome: report of 2 cases. J. Clin. Endocrinol. Metab 14, 193–204 (1954). - PubMed
-
- Seip M Lipodystrophy and gigantism with associated endocrine manifestations: a new diencephalic syndrome? Acta Paediatrica 48, 555–574 (1959). - PubMed
-
- Wiedemann HR Newly recognized congenital progeroid disorder. Am. J. Med. Genet 42, 857 (1992). - PubMed
-
- Rautenstrauch T, Snigula F & Wiedemann HR Neonatal progeroid syndrome (Wiedemann-Rautenstrauch). A follow-up study [German]. Klin. Padiatr 206, 440–443 (1994). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
