

Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges
- PMID: 26241027
- PMCID: PMC4684426
- DOI: 10.1016/j.trsl.2015.07.002
Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges
Abstract
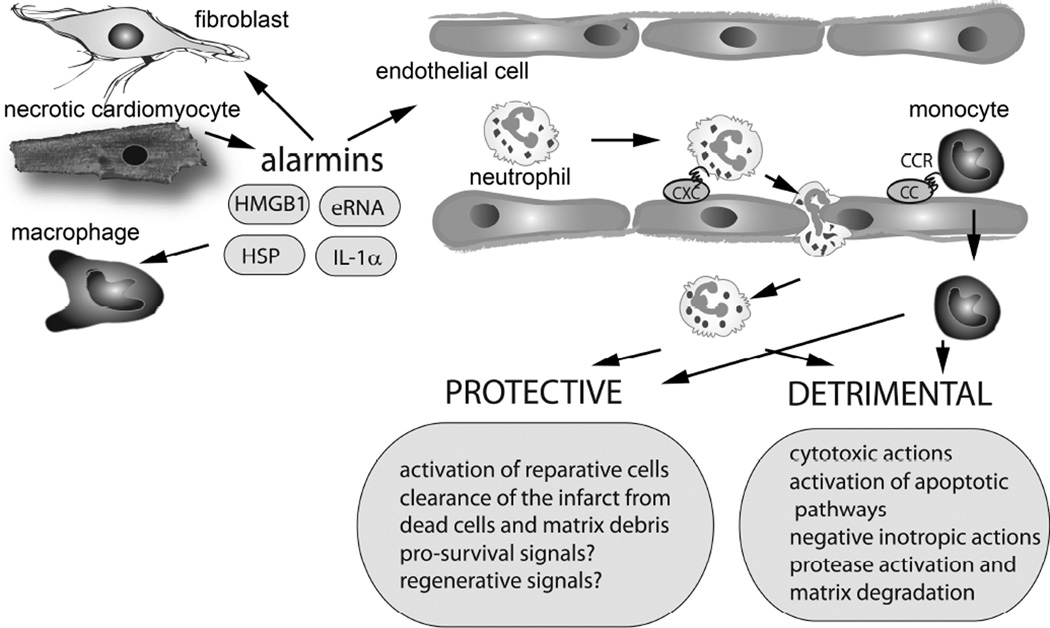
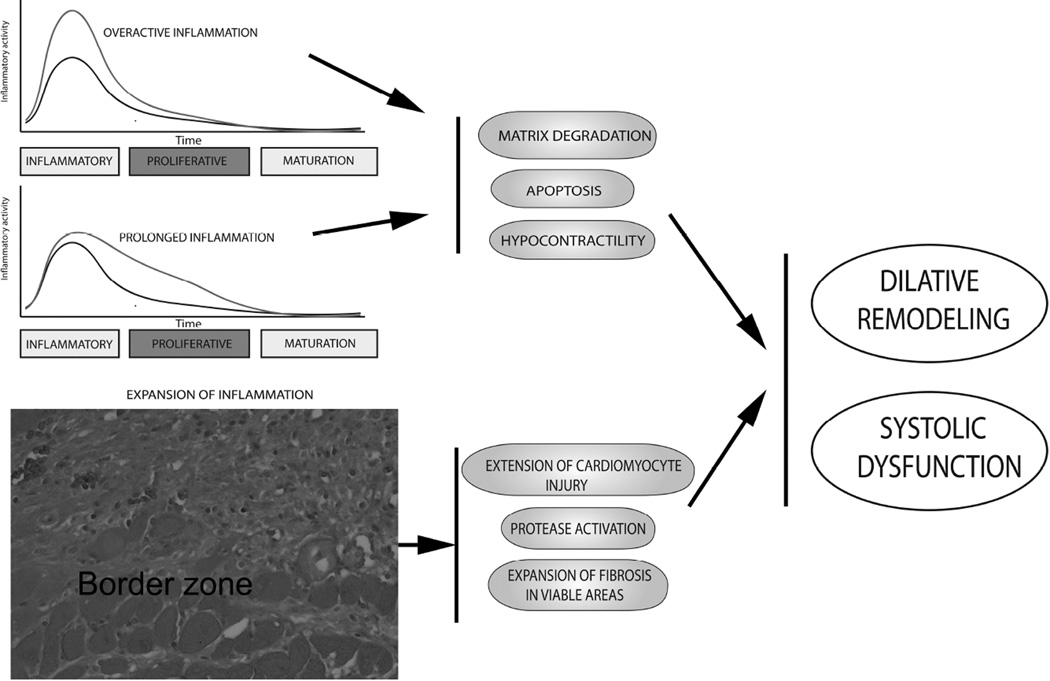
In the infarcted myocardium, necrotic cardiomyocytes release danger signals, activating an intense inflammatory response. Inflammatory pathways play a crucial role in regulation of a wide range of cellular processes involved in injury, repair, and remodeling of the infarcted heart. Proinflammatory cytokines, such as tumor necrosis factor α and interleukin 1, are markedly upregulated in the infarcted myocardium and promote adhesive interactions between endothelial cells and leukocytes by stimulating chemokine and adhesion molecule expression. Distinct pairs of chemokines and chemokine receptors are implicated in recruitment of various leukocyte subpopulations in the infarcted myocardium. For more than the past 30 years, extensive experimental work has explored the role of inflammatory signals and the contributions of leukocyte subpopulations in myocardial infarction. Robust evidence derived from experimental models of myocardial infarction has identified inflammatory targets that may attenuate cardiomyocyte injury or protect from adverse remodeling. Unfortunately, attempts to translate the promising experimental findings to clinical therapy have failed. This review article discusses the biology of the inflammatory response after myocardial infarction, attempts to identify the causes for the translational failures of the past, and proposes promising new therapeutic directions. Because of their potential involvement in injurious, reparative, and regenerative responses, inflammatory cells may hold the key for design of new therapies in myocardial infarction.
Copyright © 2016 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors have no conflicts to disclose. All authors have reviewed and approved the manuscript, and have read the journal's policy on disclosure of potential conflicts of interest and the journal's authorship agreement.
Figures
References
-
- Faxon DP, Gibbons RJ, Chronos NA, Gurbel PA, Sheehan F. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol. 2002;40:1199–1204. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
