

Double-Edge Sword of Sustained ROCK Activation in Prion Diseases through Neuritogenesis Defects and Prion Accumulation
- PMID: 26241960
- PMCID: PMC4524729
- DOI: 10.1371/journal.ppat.1005073
Double-Edge Sword of Sustained ROCK Activation in Prion Diseases through Neuritogenesis Defects and Prion Accumulation
Abstract
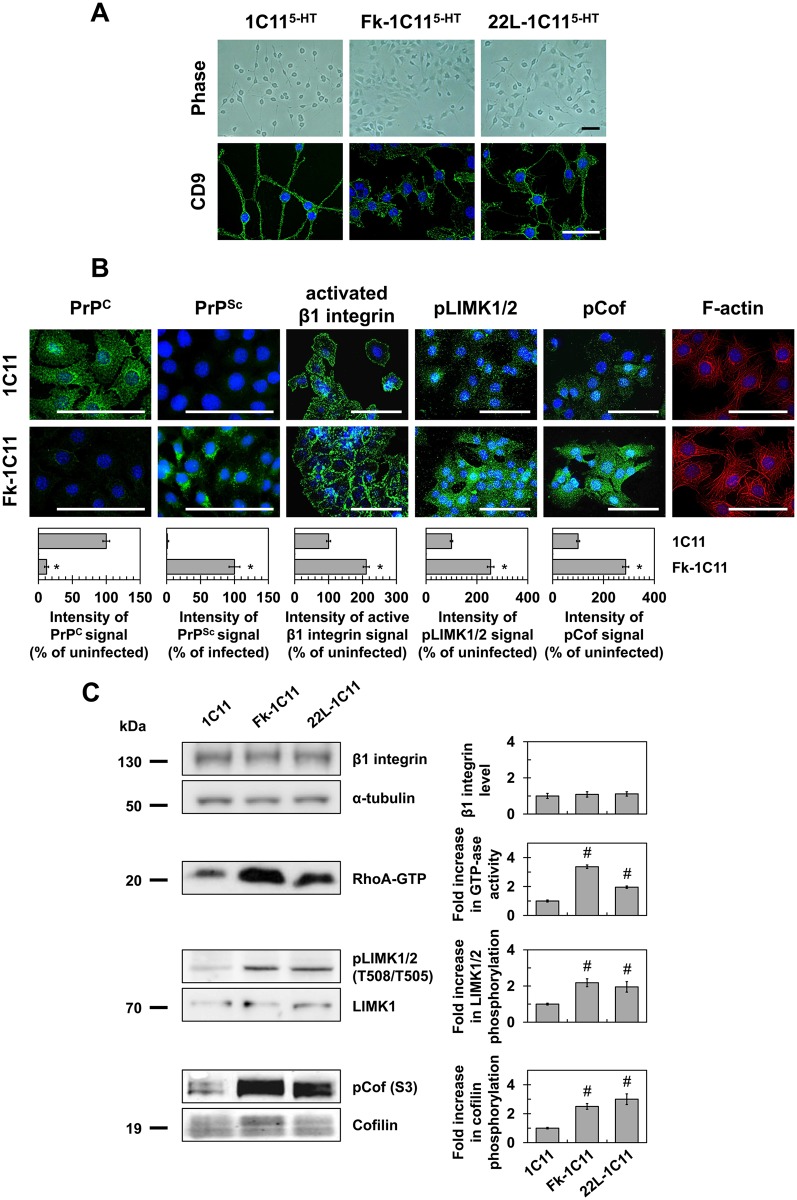
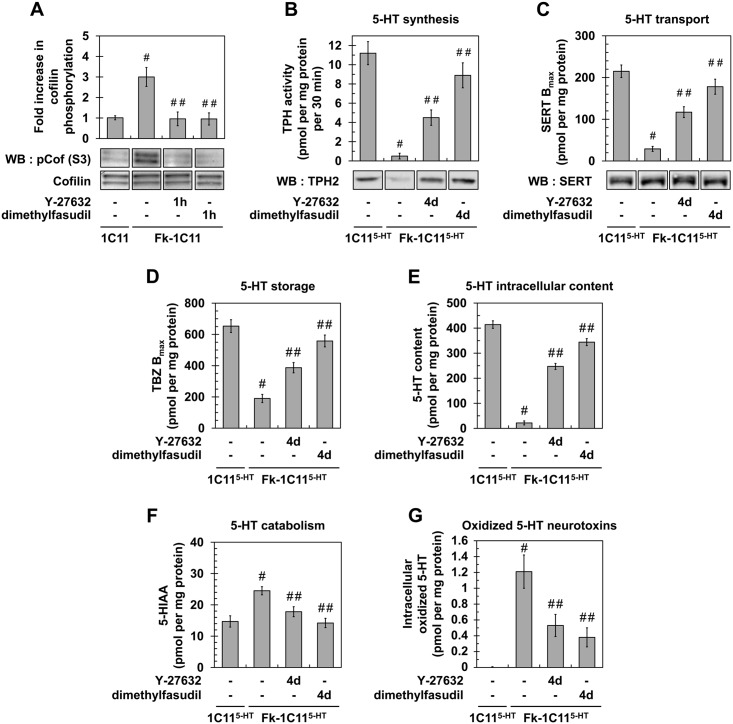
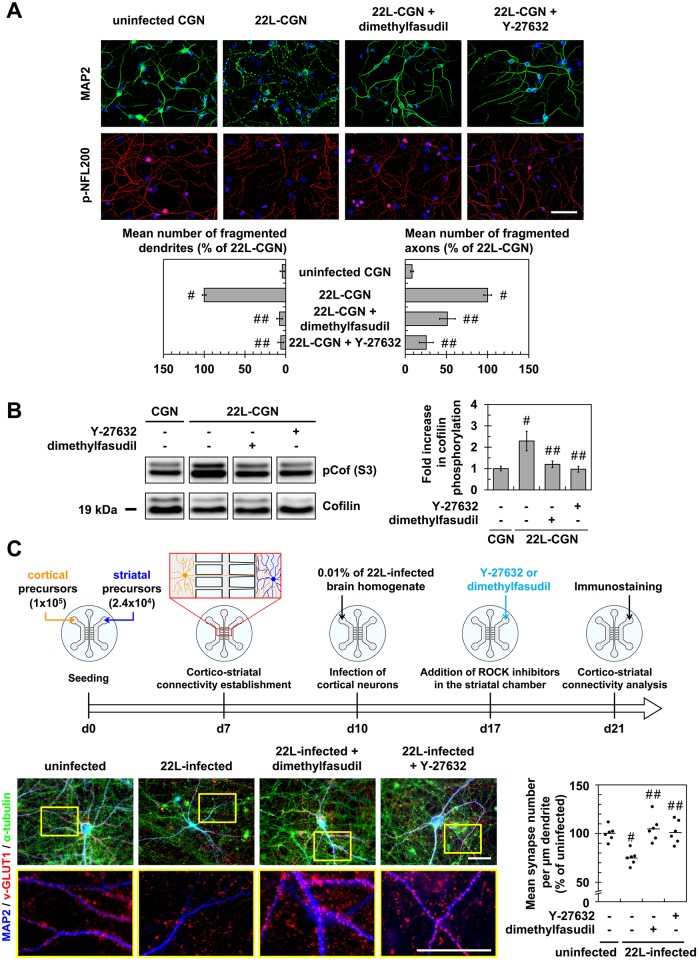
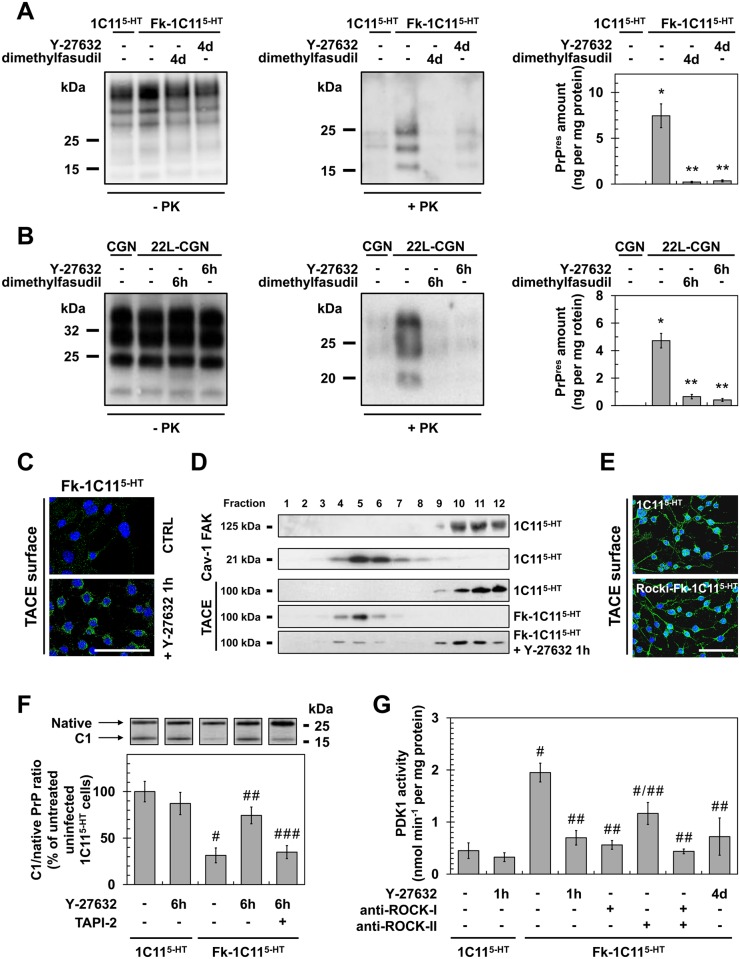
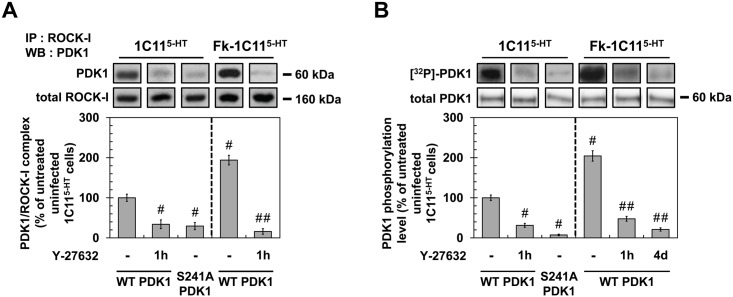
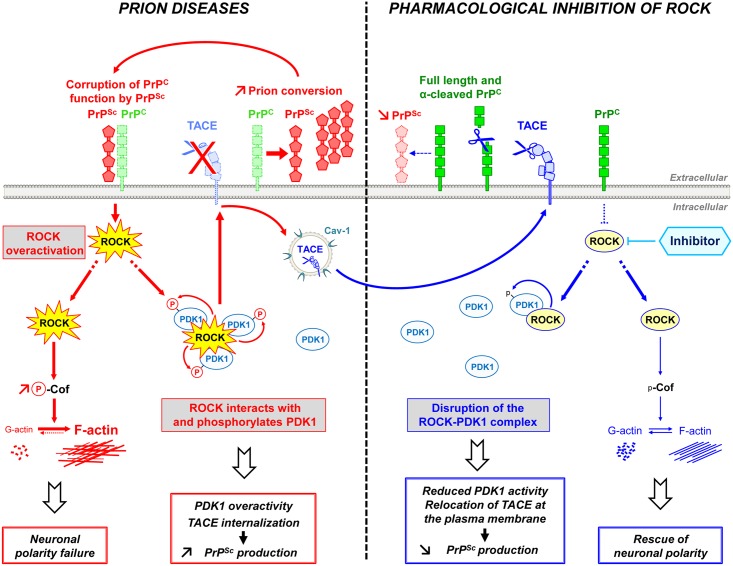
In prion diseases, synapse dysfunction, axon retraction and loss of neuronal polarity precede neuronal death. The mechanisms driving such polarization defects, however, remain unclear. Here, we examined the contribution of RhoA-associated coiled-coil containing kinases (ROCK), key players in neuritogenesis, to prion diseases. We found that overactivation of ROCK signaling occurred in neuronal stem cells infected by pathogenic prions (PrPSc) and impaired the sprouting of neurites. In reconstructed networks of mature neurons, PrPSc-induced ROCK overactivation provoked synapse disconnection and dendrite/axon degeneration. This overactivation of ROCK also disturbed overall neurotransmitter-associated functions. Importantly, we demonstrated that beyond its impact on neuronal polarity ROCK overactivity favored the production of PrPSc through a ROCK-dependent control of 3-phosphoinositide-dependent kinase 1 (PDK1) activity. In non-infectious conditions, ROCK and PDK1 associated within a complex and ROCK phosphorylated PDK1, conferring basal activity to PDK1. In prion-infected neurons, exacerbated ROCK activity increased the pool of PDK1 molecules physically interacting with and phosphorylated by ROCK. ROCK-induced PDK1 overstimulation then canceled the neuroprotective α-cleavage of normal cellular prion protein PrPC by TACE α-secretase, which physiologically precludes PrPSc production. In prion-infected cells, inhibition of ROCK rescued neurite sprouting, preserved neuronal architecture, restored neuronal functions and reduced the amount of PrPSc. In mice challenged with prions, inhibition of ROCK also lowered brain PrPSc accumulation, reduced motor impairment and extended survival. We conclude that ROCK overactivation exerts a double detrimental effect in prion diseases by altering neuronal polarity and triggering PrPSc accumulation. Eventually ROCK emerges as therapeutic target to combat prion diseases.
Conflict of interest statement
I have read the journal's policy and the authors of this manuscript have the following competing interests: JML has non-financial competing interests with Hoffmann La Roche Ltd laboratories. He acts as an expert witness for Hoffmann La Roche Ltd laboratories. This does not alter our adherence to all PLOS Pathogens policies on sharing data and materials.
Figures

References
-
- Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC (2003) Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol 13: 669–673. - PubMed
-
- Sievers C, Platt N, Perry VH, Coleman MP, Conforti L (2003) Neurites undergoing Wallerian degeneration show an apoptotic-like process with Annexin V positive staining and loss of mitochondrial membrane potential. Neurosci Res 46: 161–169. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
