

Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation
- PMID: 26245380
- PMCID: PMC4830470
- DOI: 10.1038/nature14884
Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation
Abstract
Protein aggregates are the hallmark of stressed and ageing cells, and characterize several pathophysiological states. Healthy metazoan cells effectively eliminate intracellular protein aggregates, indicating that efficient disaggregation and/or degradation mechanisms exist. However, metazoans lack the key heat-shock protein disaggregase HSP100 of non-metazoan HSP70-dependent protein disaggregation systems, and the human HSP70 system alone, even with the crucial HSP110 nucleotide exchange factor, has poor disaggregation activity in vitro. This unresolved conundrum is central to protein quality control biology. Here we show that synergic cooperation between complexed J-protein co-chaperones of classes A and B unleashes highly efficient protein disaggregation activity in human and nematode HSP70 systems. Metazoan mixed-class J-protein complexes are transient, involve complementary charged regions conserved in the J-domains and carboxy-terminal domains of each J-protein class, and are flexible with respect to subunit composition. Complex formation allows J-proteins to initiate transient higher order chaperone structures involving HSP70 and interacting nucleotide exchange factors. A network of cooperative class A and B J-protein interactions therefore provides the metazoan HSP70 machinery with powerful, flexible, and finely regulatable disaggregase activity and a further level of regulation crucial for cellular protein quality control.
Figures
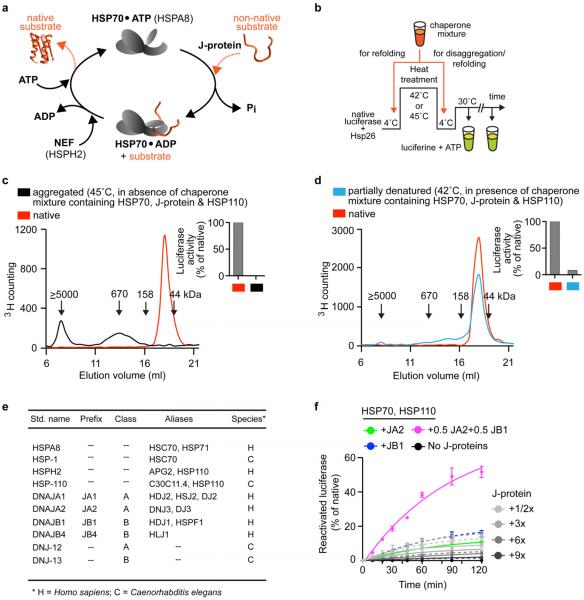
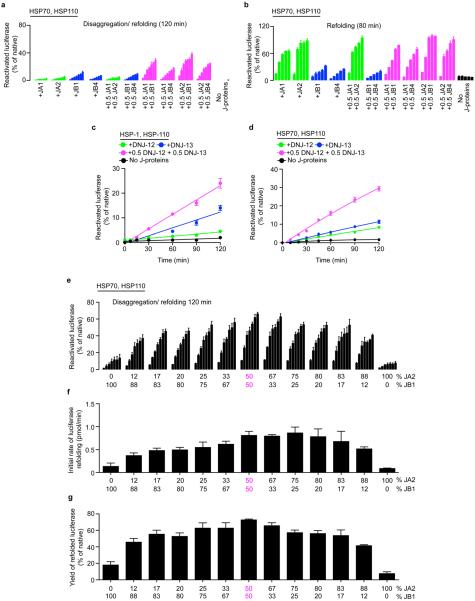
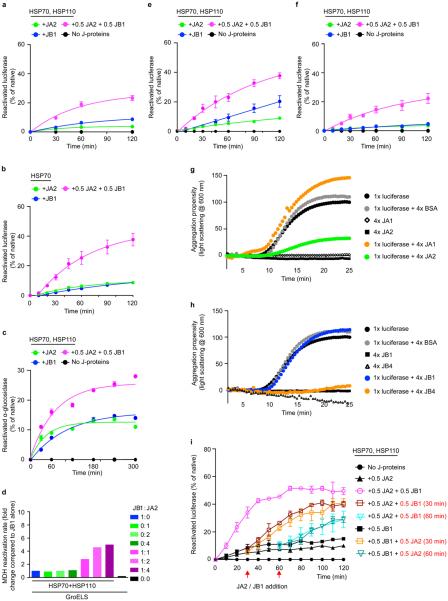
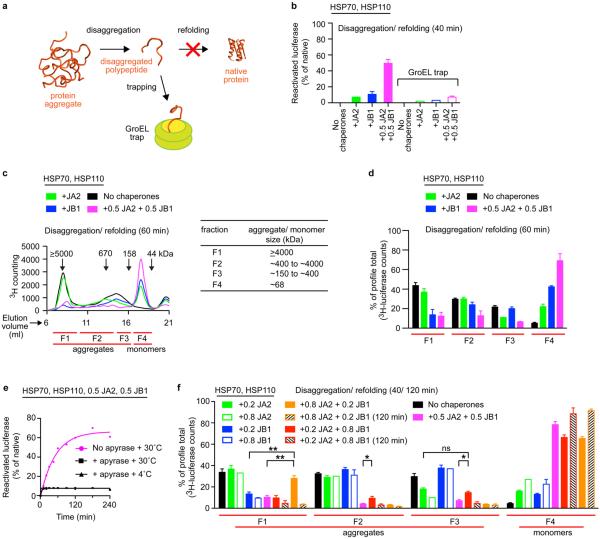
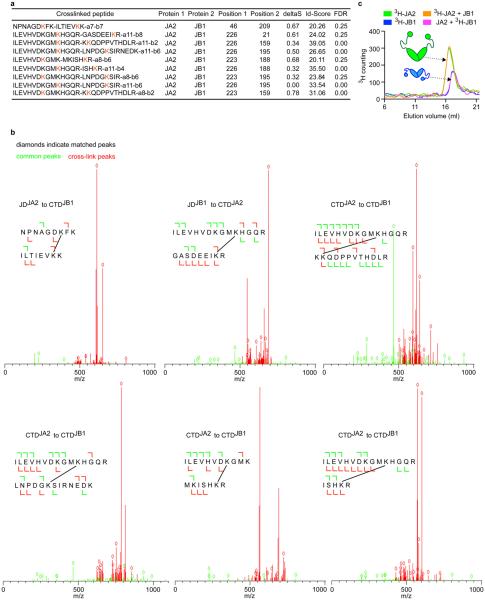
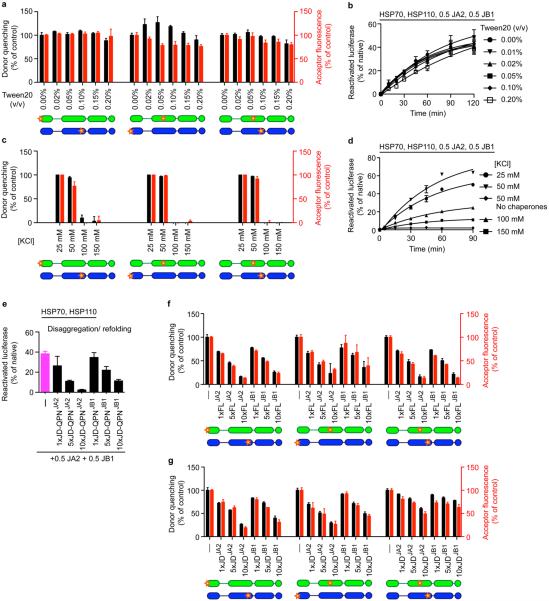
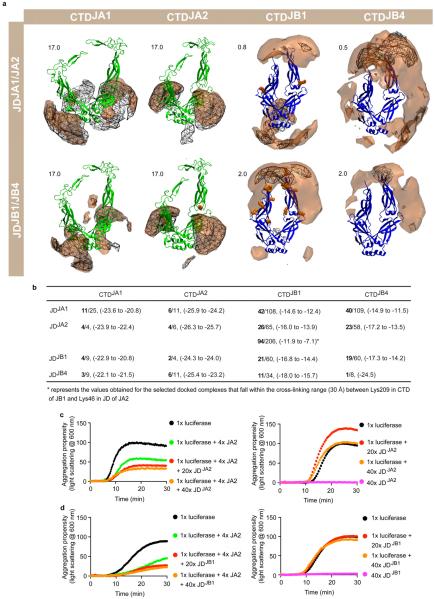
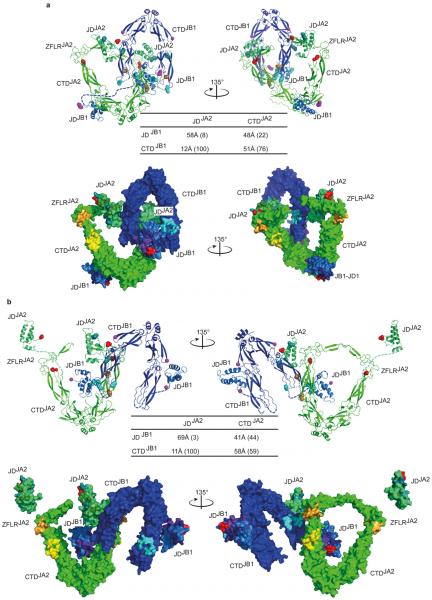
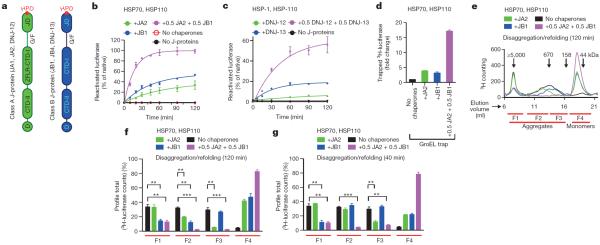
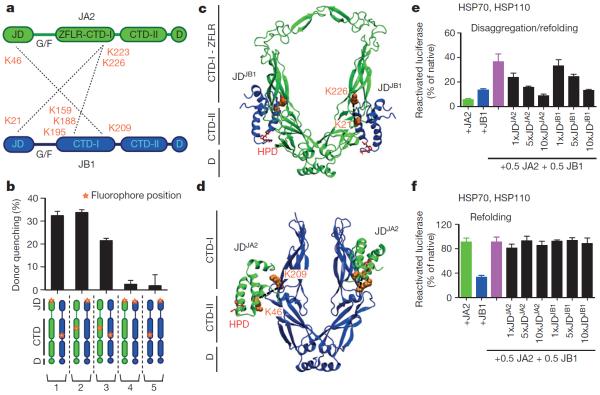
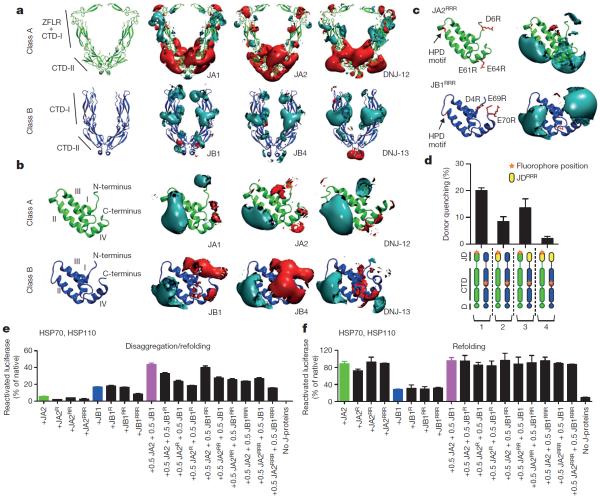
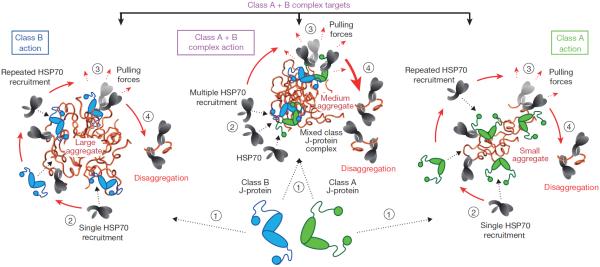
Comment in
-
Molecular biology: It takes two to untangle.Nature. 2015 Aug 13;524(7564):169-70. doi: 10.1038/nature14640. Epub 2015 Aug 5. Nature. 2015. PMID: 26245378 No abstract available.
References
-
- Hipp MS, Park SH, Hartl FU. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014;24:506–514. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
