

Host Determinants of Prion Strain Diversity Independent of Prion Protein Genotype
- PMID: 26246570
- PMCID: PMC4580196
- DOI: 10.1128/JVI.01586-15
Host Determinants of Prion Strain Diversity Independent of Prion Protein Genotype
Abstract
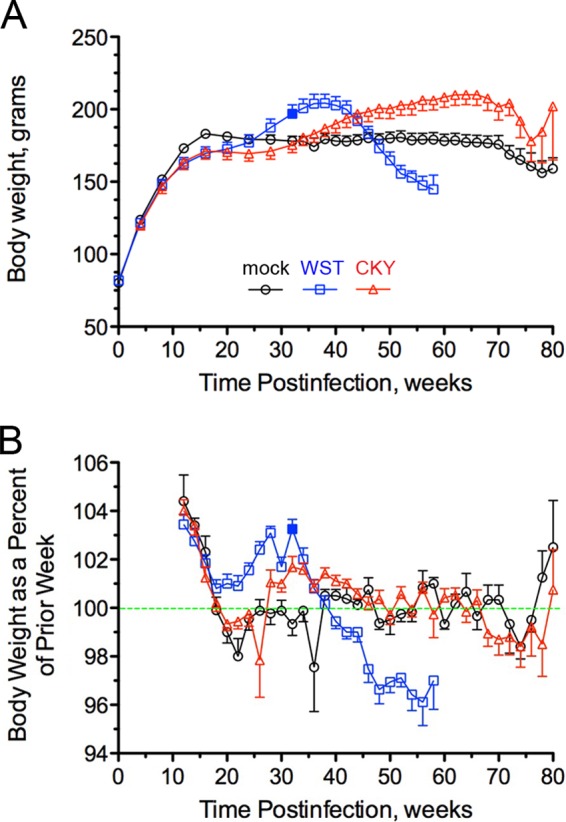
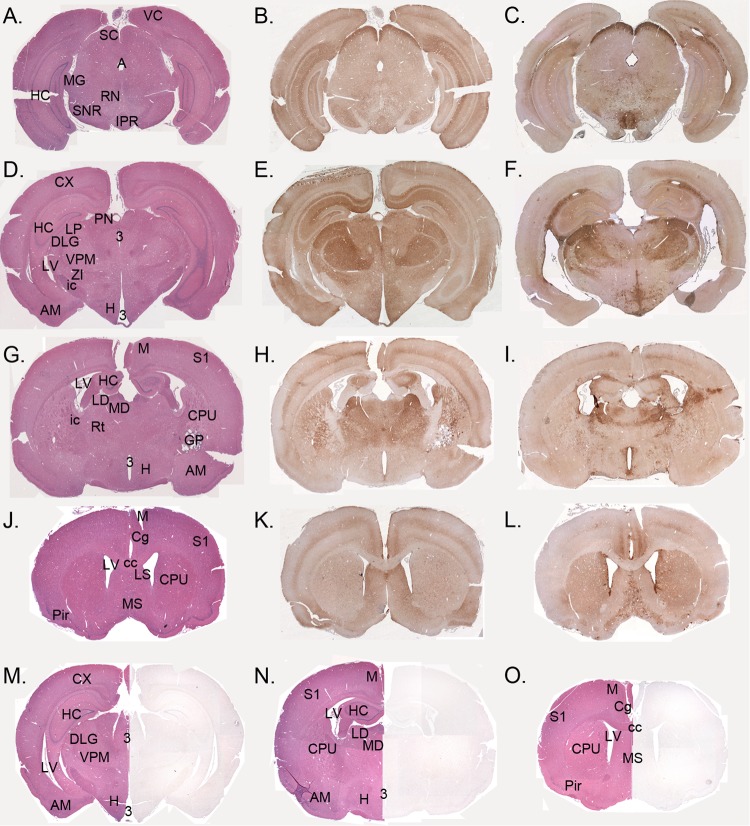
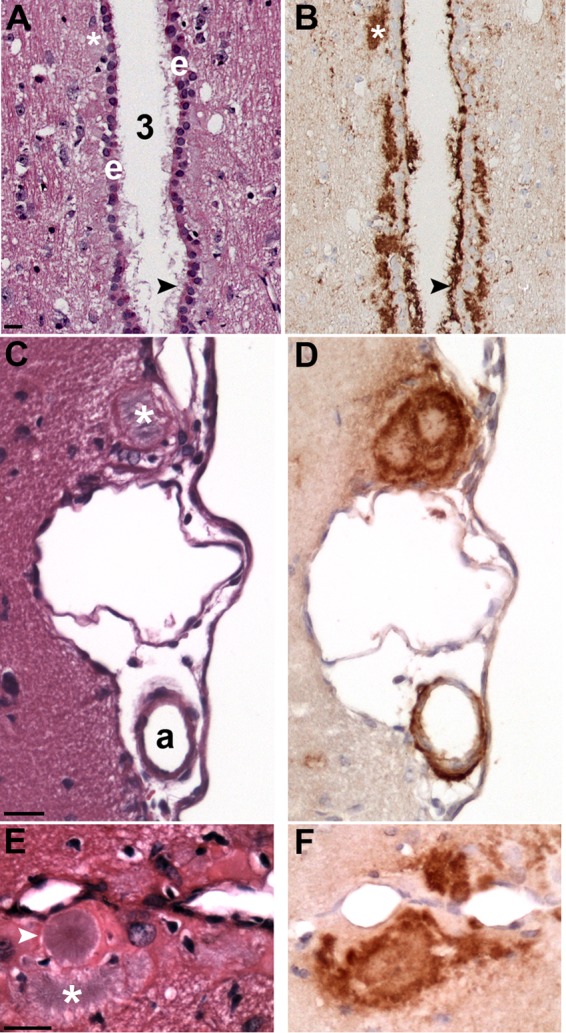
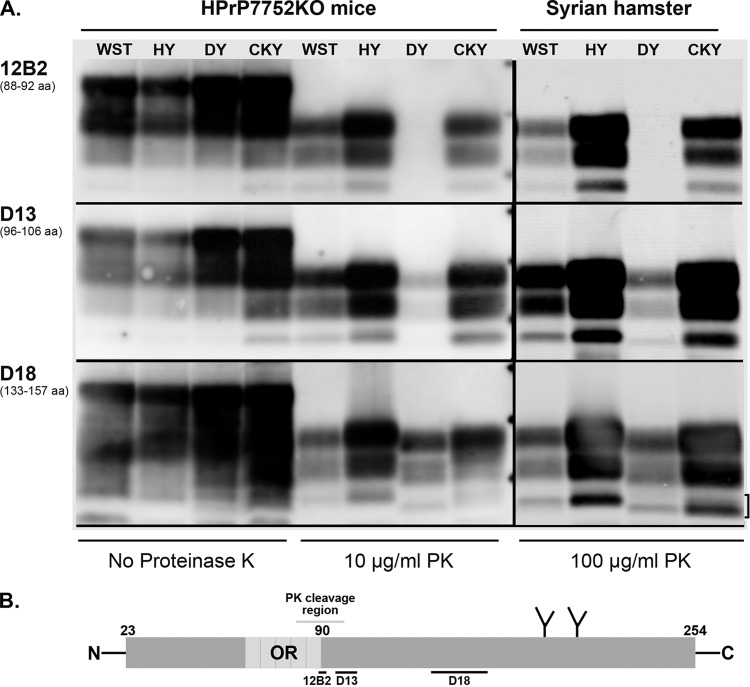
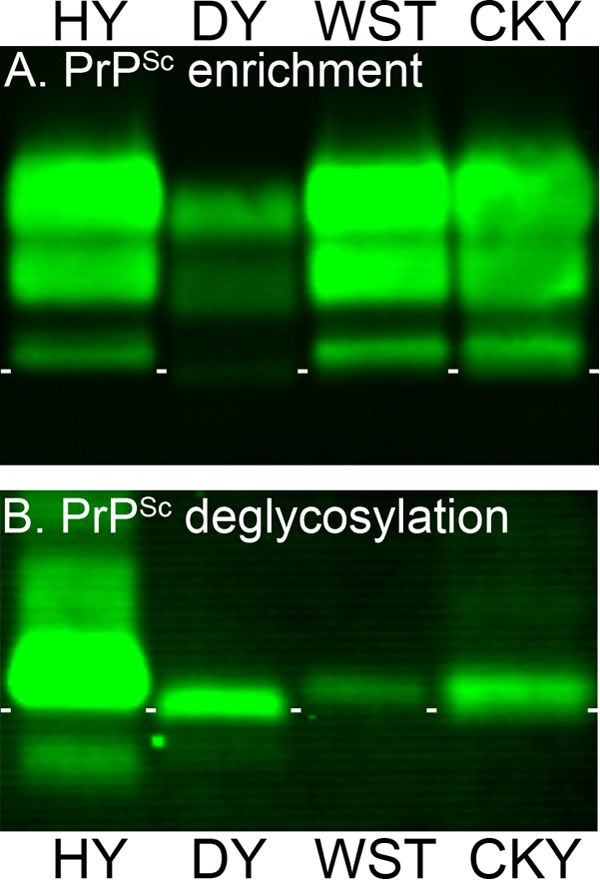
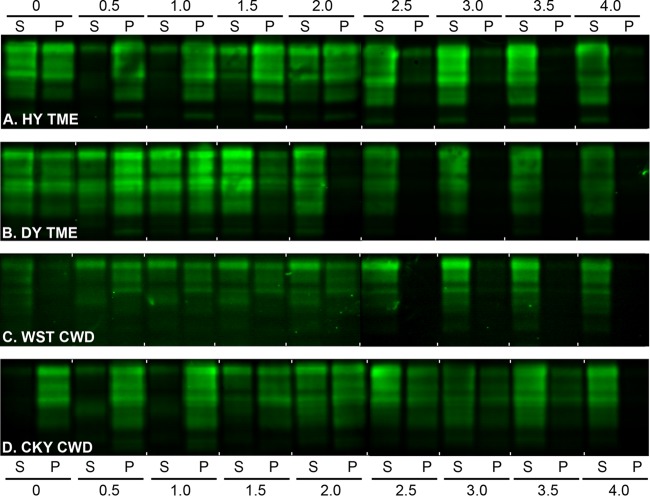
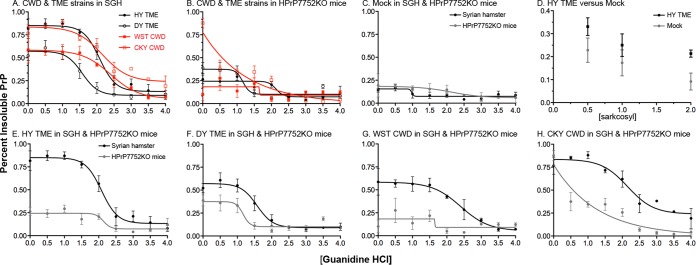
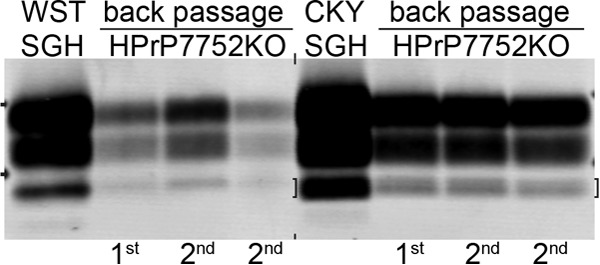
Phenotypic diversity in prion diseases can be specified by prion strains in which biological traits are propagated through an epigenetic mechanism mediated by distinct PrP(Sc) conformations. We investigated the role of host-dependent factors on phenotypic diversity of chronic wasting disease (CWD) in different host species that express the same prion protein gene (Prnp). Two CWD strains that have distinct biological, biochemical, and pathological features were identified in transgenic mice that express the Syrian golden hamster (SGH) Prnp. The CKY strain of CWD had a shorter incubation period than the WST strain of CWD, but after transmission to SGH, the incubation period of CKY CWD was ∼150 days longer than WST CWD. Limited proteinase K digestion revealed strain-specific PrP(Sc) polypeptide patterns that were maintained in both hosts, but the solubility and conformational stability of PrP(Sc) differed for the CWD strains in a host-dependent manner. WST CWD produced PrP(Sc) amyloid plaques in the brain of the SGH that were partially insoluble and stable at a high concentration of protein denaturant. However, in transgenic mice, PrP(Sc) from WST CWD did not assemble into plaques, was highly soluble, and had low conformational stability. Similar studies using the HY and DY strains of transmissible mink encephalopathy resulted in minor differences in prion biological and PrP(Sc) properties between transgenic mice and SGH. These findings indicate that host-specific pathways that are independent of Prnp can alter the PrP(Sc) conformation of certain prion strains, leading to changes in the biophysical properties of PrP(Sc), neuropathology, and clinical prion disease.
Importance: Prions are misfolded pathogenic proteins that cause neurodegeneration in humans and animals. Transmissible prion diseases exhibit a spectrum of disease phenotypes and the basis of this diversity is encoded in the structure of the pathogenic prion protein and propagated by an epigenetic mechanism. In the present study, we investigated prion diversity in two hosts species that express the same prion protein gene. While prior reports have demonstrated that prion strain properties are stable upon infection of the same host species and prion protein genotype, our findings indicate that certain prion strains can undergo dramatic changes in biological properties that are not dependent on the prion protein. Therefore, host factors independent of the prion protein can affect prion diversity. Understanding how host pathways can modify prion disease phenotypes may provide clues on how to alter prion formation and lead to treatments for prion, and other, human neurodegenerative diseases of protein misfolding.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Deer Prion Proteins Modulate the Emergence and Adaptation of Chronic Wasting Disease Strains.J Virol. 2015 Dec;89(24):12362-73. doi: 10.1128/JVI.02010-15. Epub 2015 Sep 30. J Virol. 2015. PMID: 26423950 Free PMC article.
-
Pathologic and biochemical characterization of PrPSc from elk with PRNP polymorphisms at codon 132 after experimental infection with the chronic wasting disease agent.BMC Vet Res. 2018 Mar 9;14(1):80. doi: 10.1186/s12917-018-1400-9. BMC Vet Res. 2018. PMID: 29523205 Free PMC article.
-
Raccoons accumulate PrPSc after intracranial inoculation of the agents of chronic wasting disease or transmissible mink encephalopathy but not atypical scrapie.J Vet Diagn Invest. 2019 Mar;31(2):200-209. doi: 10.1177/1040638718825290. Epub 2019 Jan 29. J Vet Diagn Invest. 2019. PMID: 30694116 Free PMC article.
-
Insights into Mechanisms of Transmission and Pathogenesis from Transgenic Mouse Models of Prion Diseases.Methods Mol Biol. 2017;1658:219-252. doi: 10.1007/978-1-4939-7244-9_16. Methods Mol Biol. 2017. PMID: 28861793 Free PMC article. Review.
-
Cervid Prion Protein Polymorphisms: Role in Chronic Wasting Disease Pathogenesis.Int J Mol Sci. 2021 Feb 25;22(5):2271. doi: 10.3390/ijms22052271. Int J Mol Sci. 2021. PMID: 33668798 Free PMC article. Review.
Cited by
-
Destabilizing polymorphism in cervid prion protein hydrophobic core determines prion conformation and conversion efficiency.PLoS Pathog. 2017 Aug 11;13(8):e1006553. doi: 10.1371/journal.ppat.1006553. eCollection 2017 Aug. PLoS Pathog. 2017. PMID: 28800624 Free PMC article.
-
Insights into Mechanisms of Chronic Neurodegeneration.Int J Mol Sci. 2016 Jan 12;17(1):82. doi: 10.3390/ijms17010082. Int J Mol Sci. 2016. PMID: 26771599 Free PMC article. Review.
-
Tissue-specific biochemical differences between chronic wasting disease prions isolated from free-ranging white-tailed deer (Odocoileus virginianus).J Biol Chem. 2022 Apr;298(4):101834. doi: 10.1016/j.jbc.2022.101834. Epub 2022 Mar 16. J Biol Chem. 2022. PMID: 35304100 Free PMC article.
-
Propagation of PrPSc in mice reveals impact of aggregate composition on prion disease pathogenesis.Commun Biol. 2023 Nov 14;6(1):1162. doi: 10.1038/s42003-023-05541-3. Commun Biol. 2023. PMID: 37964018 Free PMC article.
-
The ecology of chronic wasting disease in wildlife.Biol Rev Camb Philos Soc. 2020 Apr;95(2):393-408. doi: 10.1111/brv.12568. Epub 2019 Nov 21. Biol Rev Camb Philos Soc. 2020. PMID: 31750623 Free PMC article. Review.
References
-
- Owen F, Poulter M, Lofthouse R, Collinge J, Crow TJ, Risby D, Baker HF, Ridley RM, Hsiao K, Prusiner SB. 1989. Insertion in prion protein gene in familial Creutzfeldt-Jakob disease. Lancet i:51–52. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
