

Comparative Immunogenicity of a Cytotoxic T Cell Epitope Delivered by Penetratin and TAT Cell Penetrating Peptides
- PMID: 26247926
- PMCID: PMC6332296
- DOI: 10.3390/molecules200814033
Comparative Immunogenicity of a Cytotoxic T Cell Epitope Delivered by Penetratin and TAT Cell Penetrating Peptides
Abstract
Cell penetrating peptides (CPP), including the TAT peptide from the human immunodeficiency virus transactivator of transcription (HIV-TAT) protein and penetratin from Drosophila Antennapedia homeodomain protein, translocate various cargos including peptides and proteins across cellular barriers. This mode of delivery has been harnessed by our group and others to deliver antigenic proteins or peptides into the cytoplasm of antigen processing cells (APC) such as monocyte-derived dendritic cells (MoDC). Antigens or T cell epitopes delivered by CPP into APC in vivo generate antigen-specific cytotoxic T cell and helper T cell responses in mice. Furthermore, mice immunised with these peptides or proteins are protected from a tumour challenge. The functional properties of CPP are dependent on the various cargos being delivered and the target cell type. Despite several studies demonstrating superior immunogenicity of TAT and Antp-based immunogens, none has compared the immunogenicity of antigens delivered by TAT and Antp CPP. In the current study we demonstrate that a cytotoxic T cell epitope from the mucin 1 (MUC1) tumour associated antigen, when delivered by TAT or Antp, generates identical immune responses in mice resulting in specific MUC1 T cell responses as measured by in vivo CTL assays, IFNγ ELISpot assays and prophylactic tumour protection.
Keywords: CPP; TAT; antigen delivery; antigen presentation; cytotoxic T cell epitope; immunogenicity; immunotherapy; membrane penetrating peptide; membrane translocating peptide; penetratin; vaccine.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
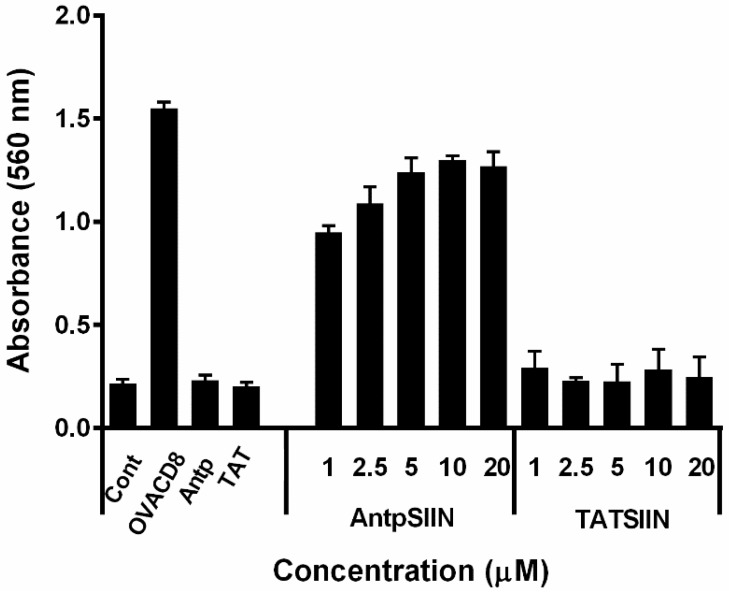
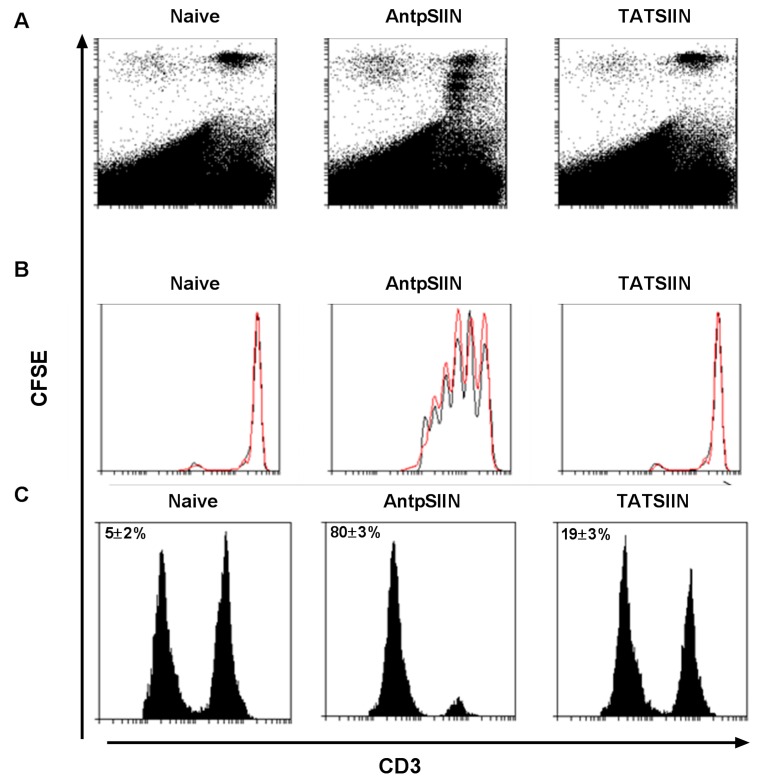
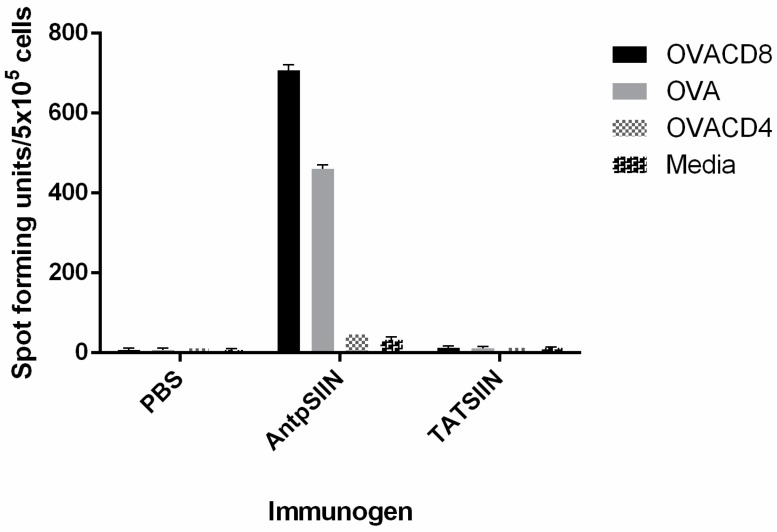
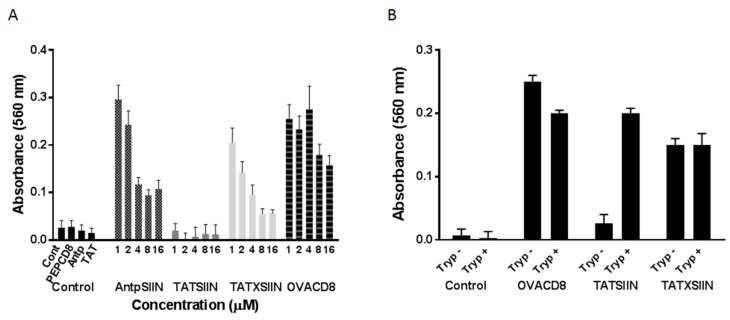
Similar articles
-
Immunogenicity of a Tripartite Cell Penetrating Peptide Containing a MUC1 Variable Number of Tandem Repeat (VNTR) and A T Helper Epitope.Molecules. 2018 Sep 2;23(9):2233. doi: 10.3390/molecules23092233. Molecules. 2018. PMID: 30200528 Free PMC article.
-
Vaccine delivery by penetratin: mechanism of antigen presentation by dendritic cells.Immunol Res. 2016 Aug;64(4):887-900. doi: 10.1007/s12026-016-8799-5. Immunol Res. 2016. PMID: 27138940
-
A membrane penetrating multiple antigen peptide (MAP) incorporating ovalbumin CD8 epitope induces potent immune responses in mice.Biochim Biophys Acta. 2010 Dec;1798(12):2286-95. doi: 10.1016/j.bbamem.2010.05.007. Epub 2010 May 15. Biochim Biophys Acta. 2010. PMID: 20478265
-
Is there anybody in there? On the mechanisms of wall crossing of cell penetrating peptides.Curr Protein Pept Sci. 2012 Nov;13(7):658-71. doi: 10.2174/138920312804142174. Curr Protein Pept Sci. 2012. PMID: 23131191 Review.
-
Cy5.5-CGRRRQRRKKRG-Labeled T lymphocytes.2008 Jul 10 [updated 2008 Aug 27]. In: Molecular Imaging and Contrast Agent Database (MICAD) [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2004–2013. 2008 Jul 10 [updated 2008 Aug 27]. In: Molecular Imaging and Contrast Agent Database (MICAD) [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2004–2013. PMID: 20641549 Free Books & Documents. Review.
Cited by
-
Intrathecal delivery of frataxin mRNA encapsulated in lipid nanoparticles to dorsal root ganglia as a potential therapeutic for Friedreich's ataxia.Sci Rep. 2016 Feb 17;6:20019. doi: 10.1038/srep20019. Sci Rep. 2016. PMID: 26883577 Free PMC article.
-
Immunogenicity of a Tripartite Cell Penetrating Peptide Containing a MUC1 Variable Number of Tandem Repeat (VNTR) and A T Helper Epitope.Molecules. 2018 Sep 2;23(9):2233. doi: 10.3390/molecules23092233. Molecules. 2018. PMID: 30200528 Free PMC article.
-
Mechanistic insights into the efficacy of cell penetrating peptide-based cancer vaccines.Cell Mol Life Sci. 2018 Aug;75(16):2887-2896. doi: 10.1007/s00018-018-2785-0. Epub 2018 Mar 5. Cell Mol Life Sci. 2018. PMID: 29508006 Free PMC article. Review.
-
Human Superantibodies to 3CLpro Inhibit Replication of SARS-CoV-2 across Variants.Int J Mol Sci. 2022 Jun 13;23(12):6587. doi: 10.3390/ijms23126587. Int J Mol Sci. 2022. PMID: 35743031 Free PMC article.
-
Cell-penetrating peptides enhance peptide vaccine accumulation and persistence in lymph nodes to drive immunogenicity.Proc Natl Acad Sci U S A. 2022 Aug 9;119(32):e2204078119. doi: 10.1073/pnas.2204078119. Epub 2022 Aug 1. Proc Natl Acad Sci U S A. 2022. PMID: 35914154 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
