

InteMAP: Integrated metagenomic assembly pipeline for NGS short reads
- PMID: 26250558
- PMCID: PMC4545859
- DOI: 10.1186/s12859-015-0686-x
InteMAP: Integrated metagenomic assembly pipeline for NGS short reads
Abstract
Background: Next-generation sequencing (NGS) has greatly facilitated metagenomic analysis but also raised new challenges for metagenomic DNA sequence assembly, owing to its high-throughput nature and extremely short reads generated by sequencers such as Illumina. To date, how to generate a high-quality draft assembly for metagenomic sequencing projects has not been fully addressed.
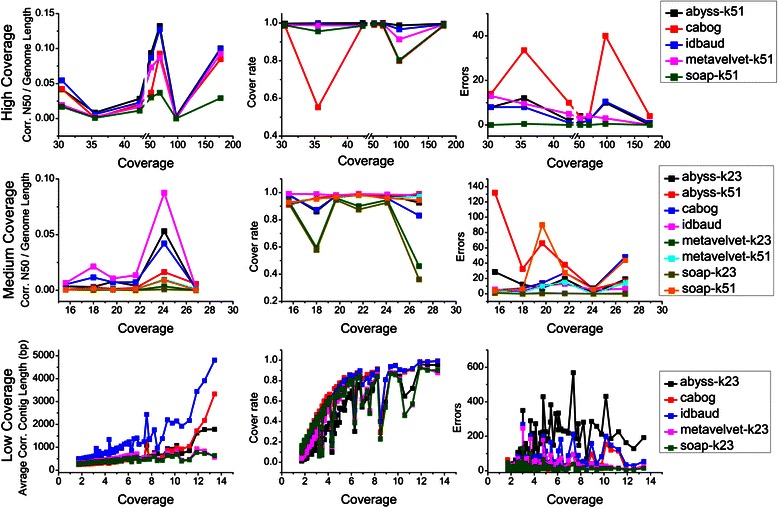
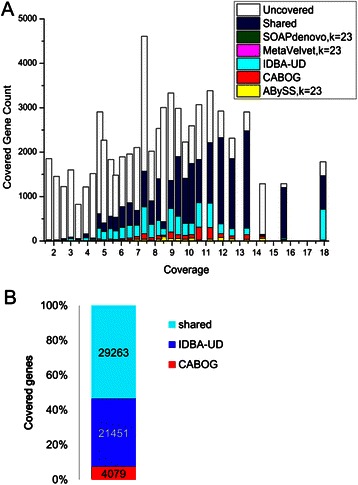
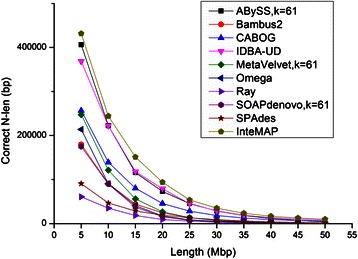
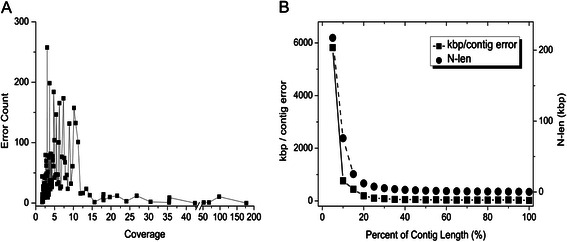
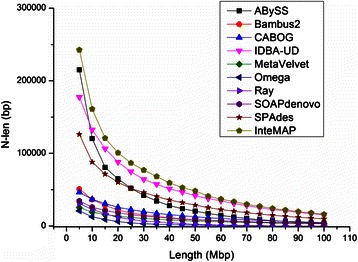
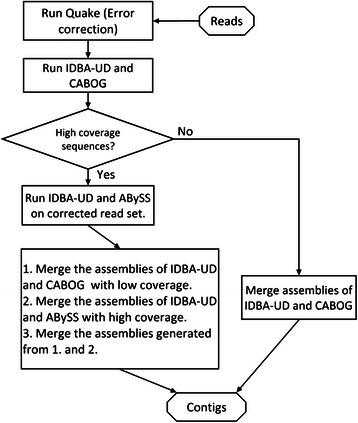
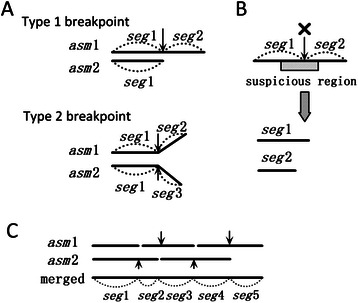
Results: We conducted a comprehensive assessment on state-of-the-art de novo assemblers and revealed that the performance of each assembler depends critically on the sequencing depth. To address this problem, we developed a pipeline named InteMAP to integrate three assemblers, ABySS, IDBA-UD and CABOG, which were found to complement each other in assembling metagenomic sequences. Making a decision of which assembling approaches to use according to the sequencing coverage estimation algorithm for each short read, the pipeline presents an automatic platform suitable to assemble real metagenomic NGS data with uneven coverage distribution of sequencing depth. By comparing the performance of InteMAP with current assemblers on both synthetic and real NGS metagenomic data, we demonstrated that InteMAP achieves better performance with a longer total contig length and higher contiguity, and contains more genes than others.
Conclusions: We developed a de novo pipeline, named InteMAP, that integrates existing tools for metagenomics assembly. The pipeline outperforms previous assembly methods on metagenomic assembly by providing a longer total contig length, a higher contiguity and covering more genes. InteMAP, therefore, could potentially be a useful tool for the research community of metagenomics.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
