

Phylogenetic analysis of methionine synthesis genes from Thalassiosira pseudonana
- PMID: 26251775
- PMCID: PMC4523565
- DOI: 10.1186/s40064-015-1163-8
Phylogenetic analysis of methionine synthesis genes from Thalassiosira pseudonana
Abstract
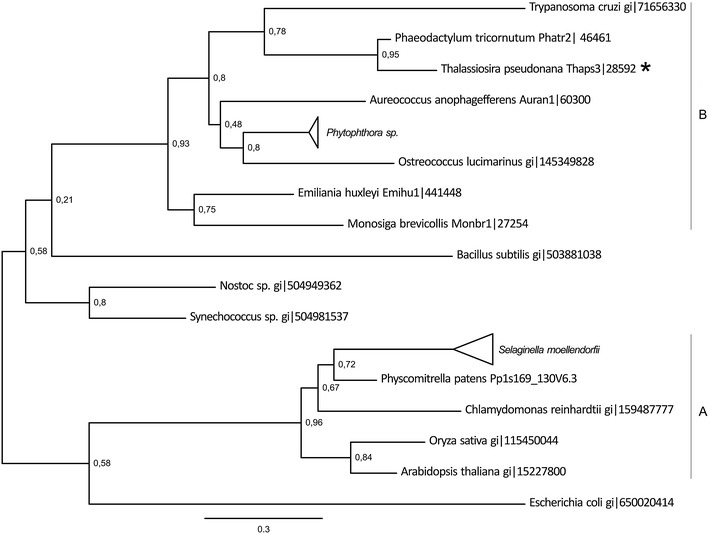
Diatoms are unicellular algae responsible for approximately 20% of global carbon fixation. Their evolution by secondary endocytobiosis resulted in a complex cellular structure and metabolism compared to algae with primary plastids. The sulfate assimilation and methionine synthesis pathways provide S-containing amino acids for the synthesis of proteins and a range of metabolites such as dimethylsulfoniopropionate. To obtain an insight into the localization and organization of the sulfur metabolism pathways we surveyed the genome of Thalassiosira pseudonana-a model organism for diatom research. We have identified and annotated genes for enzymes involved in respective pathways. Protein localization was predicted using similarities to known signal peptide motifs. We performed detailed phylogenetic analyses of enzymes involved in sulfate uptake/reduction and methionine metabolism. Moreover, we have found in up-stream sequences of studied diatoms methionine biosynthesis genes a conserved motif, which shows similarity to the Met31, a cis-motif regulating expression of methionine biosynthesis genes in yeast.
Keywords: Diatoms; Methionine synthesis; Phylogenetics.
Figures





Similar articles
-
Phylogenetic aspects of the sulfate assimilation genes from Thalassiosira pseudonana.Amino Acids. 2013 May;44(5):1253-65. doi: 10.1007/s00726-013-1462-8. Epub 2013 Jan 26. Amino Acids. 2013. PMID: 23354278 Review.
-
A model for carbohydrate metabolism in the diatom Phaeodactylum tricornutum deduced from comparative whole genome analysis.PLoS One. 2008 Jan 9;3(1):e1426. doi: 10.1371/journal.pone.0001426. PLoS One. 2008. PMID: 18183306 Free PMC article.
-
Dimethylsulfoniopropionate biosynthesis in a diatom Thalassiosira pseudonana: Identification of a gene encoding MTHB-methyltransferase.Arch Biochem Biophys. 2018 May 1;645:100-106. doi: 10.1016/j.abb.2018.03.019. Epub 2018 Mar 21. Arch Biochem Biophys. 2018. PMID: 29574051
-
Genome-scale metabolic model of the diatom Thalassiosira pseudonana highlights the importance of nitrogen and sulfur metabolism in redox balance.PLoS One. 2021 Mar 24;16(3):e0241960. doi: 10.1371/journal.pone.0241960. eCollection 2021. PLoS One. 2021. PMID: 33760840 Free PMC article.
-
Silica biomineralization in diatoms: the model organism Thalassiosira pseudonana.Chembiochem. 2008 May 23;9(8):1187-94. doi: 10.1002/cbic.200700764. Chembiochem. 2008. PMID: 18381716 Review.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
