

Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy
- PMID: 26257172
- PMCID: PMC4545408
- DOI: 10.1016/j.celrep.2015.07.023
Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy
Abstract
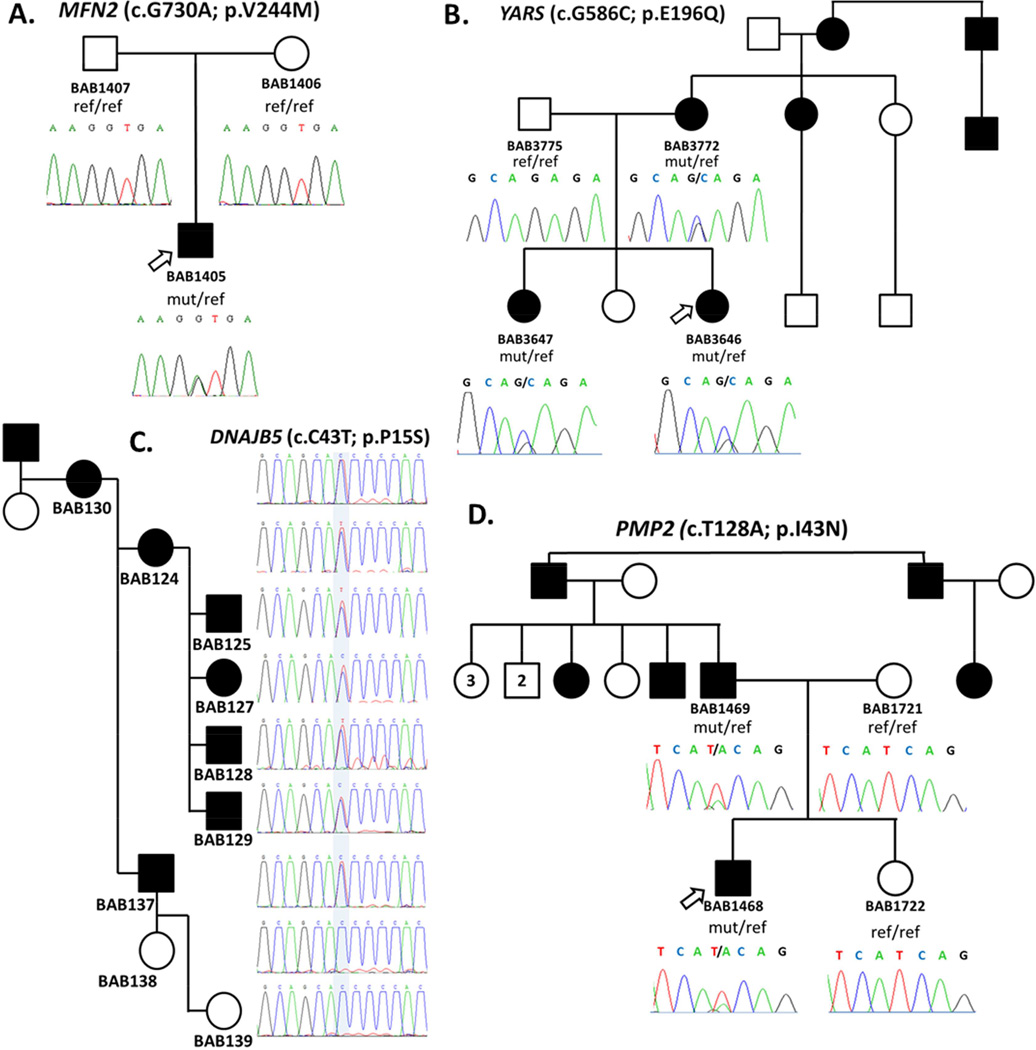
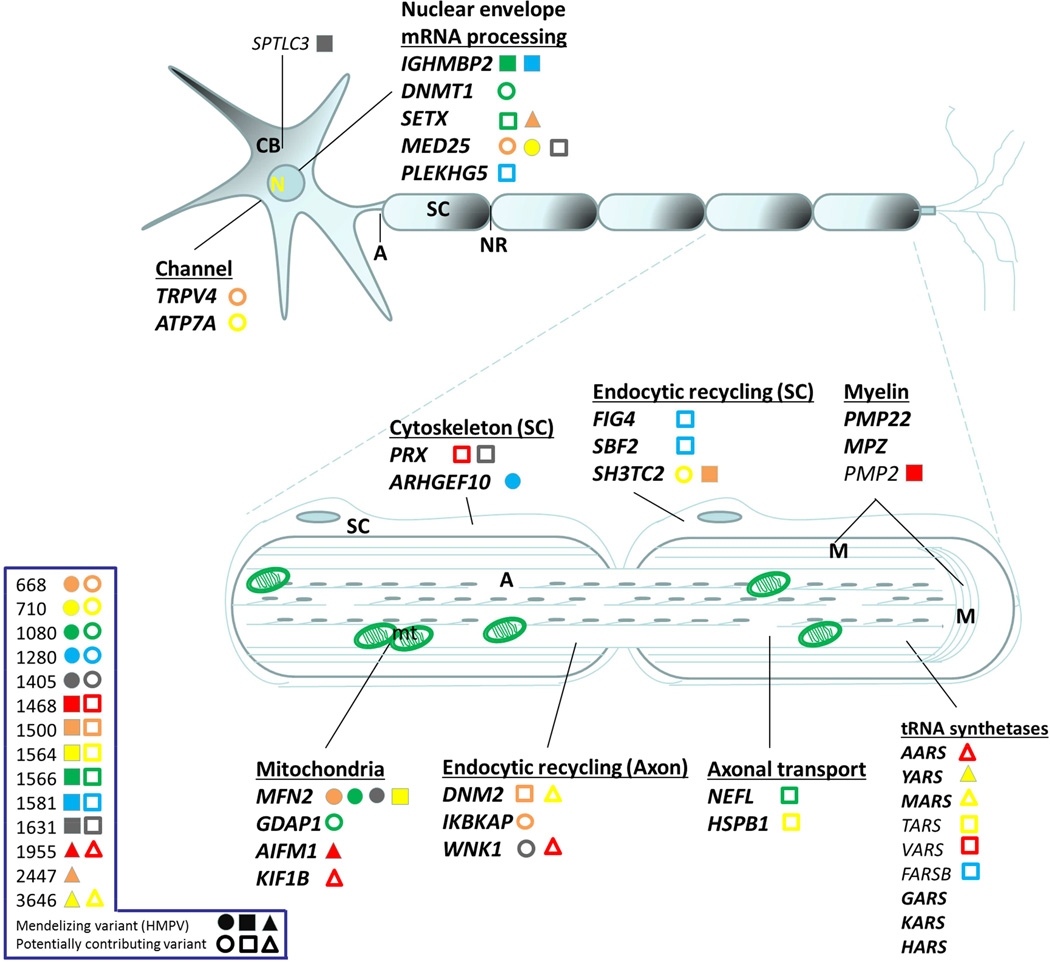
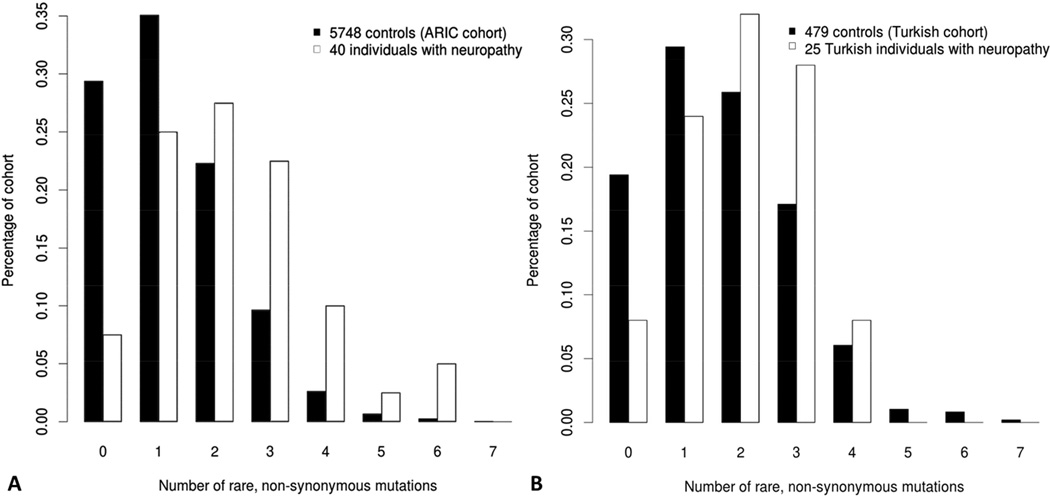
Charcot-Marie-Tooth (CMT) disease is a clinically and genetically heterogeneous distal symmetric polyneuropathy. Whole-exome sequencing (WES) of 40 individuals from 37 unrelated families with CMT-like peripheral neuropathy refractory to molecular diagnosis identified apparent causal mutations in ∼ 45% (17/37) of families. Three candidate disease genes are proposed, supported by a combination of genetic and in vivo studies. Aggregate analysis of mutation data revealed a significantly increased number of rare variants across 58 neuropathy-associated genes in subjects versus controls, confirmed in a second ethnically discrete neuropathy cohort, suggesting that mutation burden potentially contributes to phenotypic variability. Neuropathy genes shown to have highly penetrant Mendelizing variants (HPMVs) and implicated by burden in families were shown to interact genetically in a zebrafish assay exacerbating the phenotype established by the suppression of single genes. Our findings suggest that the combinatorial effect of rare variants contributes to disease burden and variable expressivity.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures


References
-
- Allan W. Relation of hereditary pattern to clinical severity as illustrated by peroneal atrophy. Arch. Intern. Med. 1939;63:1123–1131.
-
- Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N. Engl. J. Med. 2012;366(7):636–646. - PubMed
-
- Armstrong L, Biancheri R, Shyr C, Rossi A, Sinclair G, Ross CJ, Tarailo-Graovac M, Wasserman WW, van Karnebeek CD. AIMP1 deficiency presents as a cortical neurodegenerative disease with infantile onset. Neurogenetics. 2014;15(3):157–159. - PubMed
-
- Ben Othmane K, Middleton LT, Loprest LJ, Wilkinson KM, Lennon F, Rozear MP, Stajich JM, Gaskell PC, Roses AD, Pericak-Vance MA, et al. Localization of a gene (CMT2A) for autosomal dominant Charcot-Marie-Tooth disease type 2 to chromosome 1p and evidence of genetic heterogeneity. Genomics. 1993;17(2):370–375. - PubMed
Publication types
MeSH terms
Substances
Associated data
- dbGaP/SAMN03360995
- dbGaP/SAMN03361007
- dbGaP/SAMN03361017
- dbGaP/SAMN03361019
- dbGaP/SAMN03361023
- dbGaP/SAMN03361038
- dbGaP/SAMN03361050
- dbGaP/SAMN03361070
- dbGaP/SAMN03361073
- dbGaP/SAMN03361075
- dbGaP/SAMN03361077
- dbGaP/SAMN03361083
- dbGaP/SAMN03361097
- dbGaP/SAMN03361098
- dbGaP/SAMN03361130
- dbGaP/SAMN03361138
- dbGaP/SAMN03361140
- dbGaP/SAMN03361149
- dbGaP/SAMN03361168
- dbGaP/SAMN03361181
- dbGaP/SAMN03361182
- dbGaP/SAMN03361200
Grants and funding
- P50 MH094268/MH/NIMH NIH HHS/United States
- U54HG003273/HG/NHGRI NIH HHS/United States
- U54 HG006542/HG/NHGRI NIH HHS/United States
- T32 GM007526/GM/NIGMS NIH HHS/United States
- U54 NS065712/NS/NINDS NIH HHS/United States
- R01NS058529/NS/NINDS NIH HHS/United States
- U54HG006542/HG/NHGRI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- GM07863/GM/NIGMS NIH HHS/United States
- T32 GM007863/GM/NIGMS NIH HHS/United States
- K23 NS078056/NS/NINDS NIH HHS/United States
- T32 GM07526-37/GM/NIGMS NIH HHS/United States
- F30 NS092238/NS/NINDS NIH HHS/United States
- U54NS065712/NS/NINDS NIH HHS/United States
- R01 NS058529/NS/NINDS NIH HHS/United States
- GM007315/GM/NIGMS NIH HHS/United States
- K23NS078056/NS/NINDS NIH HHS/United States
- R01NS075764/NS/NINDS NIH HHS/United States
- R01 NS075764/NS/NINDS NIH HHS/United States
- NS092238/NS/NINDS NIH HHS/United States
- T32 GM007315/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
