

Safety of laronidase delivered into the spinal canal for treatment of cervical stenosis in mucopolysaccharidosis I
- PMID: 26260077
- PMCID: PMC4572891
- DOI: 10.1016/j.ymgme.2015.07.005
Safety of laronidase delivered into the spinal canal for treatment of cervical stenosis in mucopolysaccharidosis I
Abstract
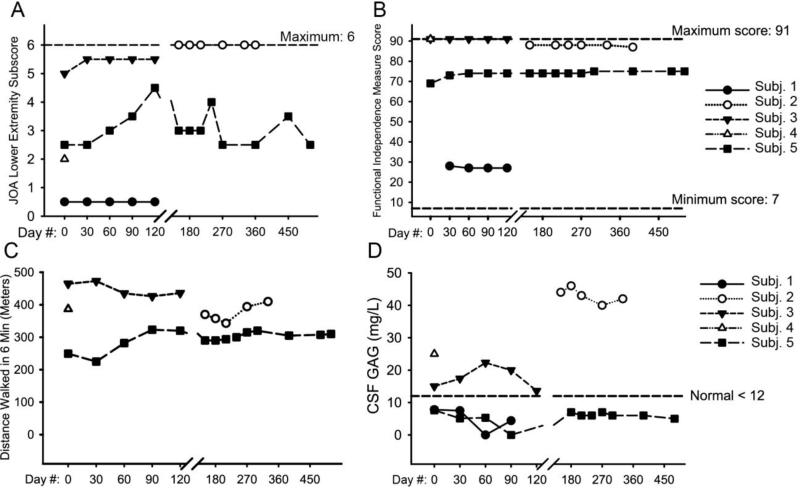
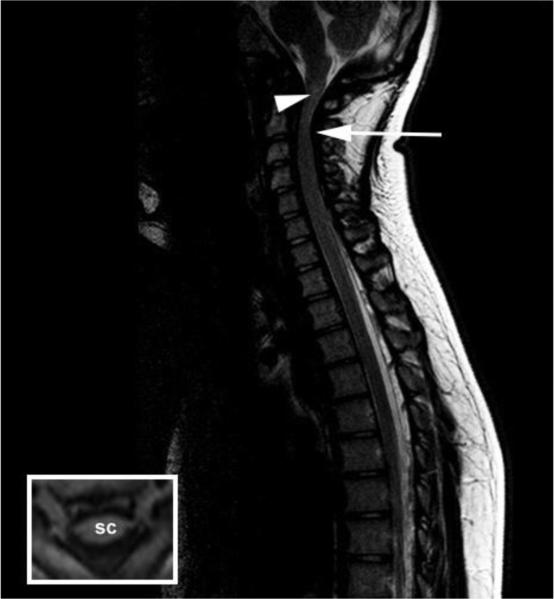
Enzyme replacement therapy with laronidase (recombinant human alpha-l-iduronidase) is successfully used to treat patients with mucopolysaccharidosis type I (MPS I). However, the intravenously-administered enzyme is not expected to treat or prevent neurological deterioration. As MPS I patients suffer from spinal cord compression due in part to thickened spinal meninges, we undertook a phase I clinical trial of lumbar intrathecal laronidase in MPS I subjects age 8 years and older with symptomatic (primarily cervical) spinal cord compression. The study faced significant challenges, including a heterogeneous patient population, difficulty recruiting subjects despite an international collaborative effort, and an inability to include a placebo-controlled design due to ethical concerns. Nine serious adverse events occurred in the subjects. All subjects reported improvement in symptomatology and showed improved neurological examinations, but objective outcome measures did not demonstrate change. Despite limitations, we demonstrated the safety of this approach to treating neurological disease due to MPS I.
Keywords: Alpha-l-iduronidase; Enzyme replacement therapy; Hurler; Intrathecal; Lysosomal storage disease; Scheie.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
References
-
- Brady RO, Pentchev PG, Gal AE, Hibbert SR, Dekaban AS. Replacement therapy for inherited enzyme deficiency: use of purified glucocerebrosidase in Gaucher's disease. N. Engl. J. Med. 1974;291:989–993. - PubMed
-
- Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Doppelt SH, Hill SC, Mankin HJ, Murray GJ, Parker RI, Argoff CE, Grewal RP, Yu KT, collaborators Replacement therapy for inherited enzyme deficiency: macrophage-targeted glucocerebrosidase for Gaucher's disease. N. Engl. J. Med. 1991;324:1464–1470. - PubMed
-
- Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ, G., the International Fabry Disease Study Safety and efficacy of recombinant human α-galactosidase A replacement therapy in Fabry's disease. N. Engl. J. Med. 2001;345:9–16. - PubMed
-
- Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J, Passage M, Izykowski B, Phillips J, Doroshow R, Walot I, Hoft R, Neufeld E. Enzyme-replacement therapy in mucopolysaccharisosis I. N. Engl. J. Med. 2001;344:182–188. - PubMed
-
- Harmatz P, Giugliani R, Schwartz I, Guffon N, Teles EL, Miranda MCS, Wraith JE, Beck M, Arash L, Scarpa M, Yu ZF, Wittes J, Berger KI, Newman MS, Lowe AM, Kakkis E, Swiedler SJ. Enzyme replacement therapy for mucopolysaccharidosis VI: A phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J. Pediatr. 2006;148:533–539. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
