

Genomes by design
- PMID: 26260262
- PMCID: PMC5484418
- DOI: 10.1038/nrg3956
Genomes by design
Abstract
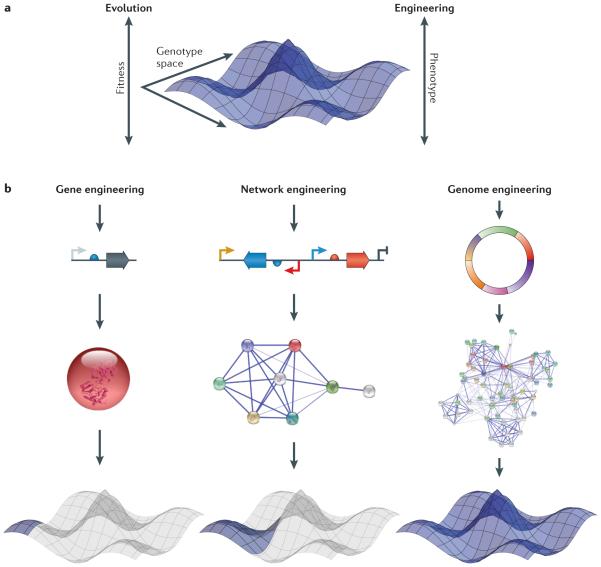
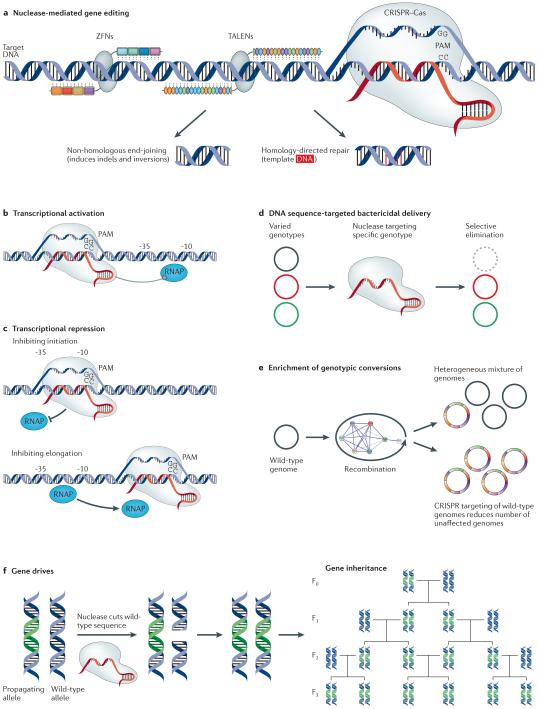
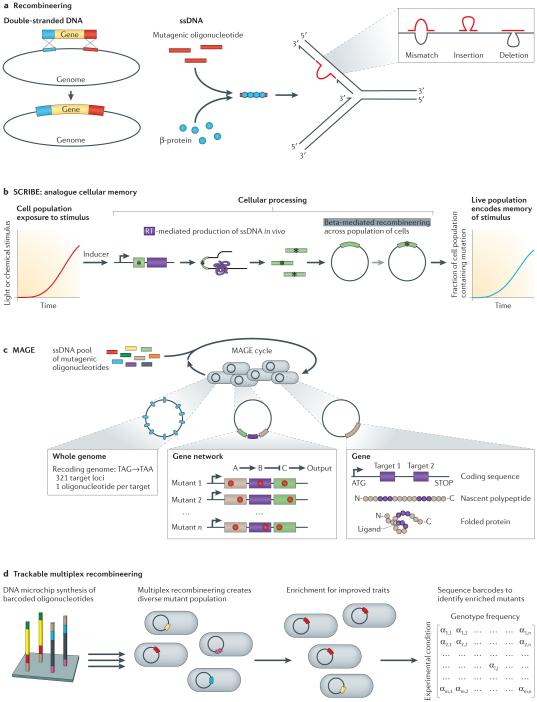
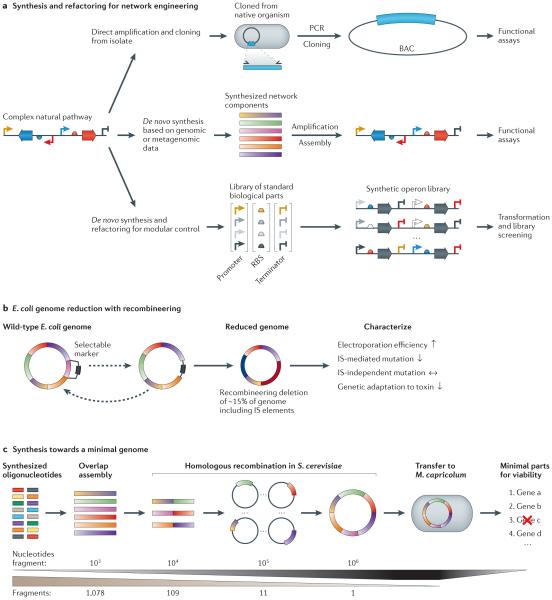
Next-generation DNA sequencing has revealed the complete genome sequences of numerous organisms, establishing a fundamental and growing understanding of genetic variation and phenotypic diversity. Engineering at the gene, network and whole-genome scale aims to introduce targeted genetic changes both to explore emergent phenotypes and to introduce new functionalities. Expansion of these approaches into massively parallel platforms establishes the ability to generate targeted genome modifications, elucidating causal links between genotype and phenotype, as well as the ability to design and reprogramme organisms. In this Review, we explore techniques and applications in genome engineering, outlining key advances and defining challenges.
Figures
References
-
- Johnson A. The Hidden Writer. Random House; 1997.
-
- Simon R, Priefer U, Puhler AA. Broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat. Biotechnol. 1983;1:784–791.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
