

Inflammation and plaque vulnerability
- PMID: 26260307
- PMCID: PMC5082111
- DOI: 10.1111/joim.12406
Inflammation and plaque vulnerability
Abstract
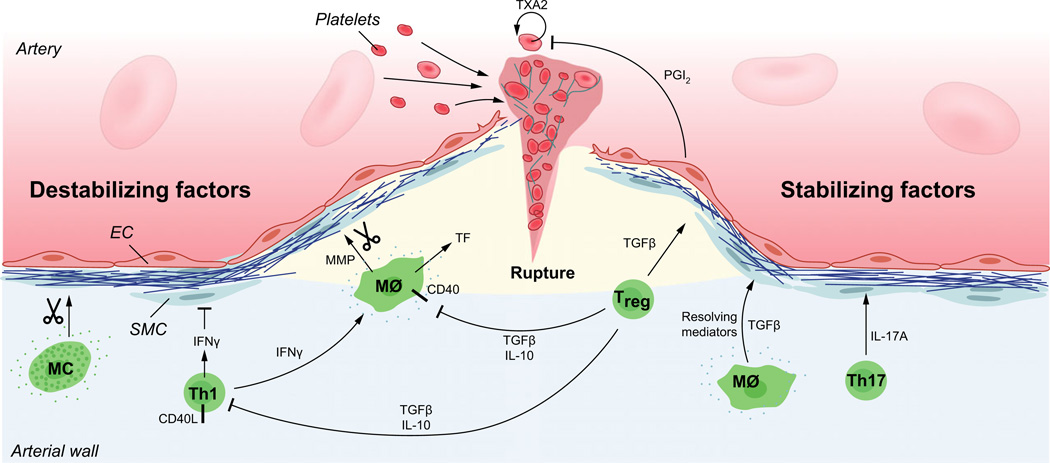
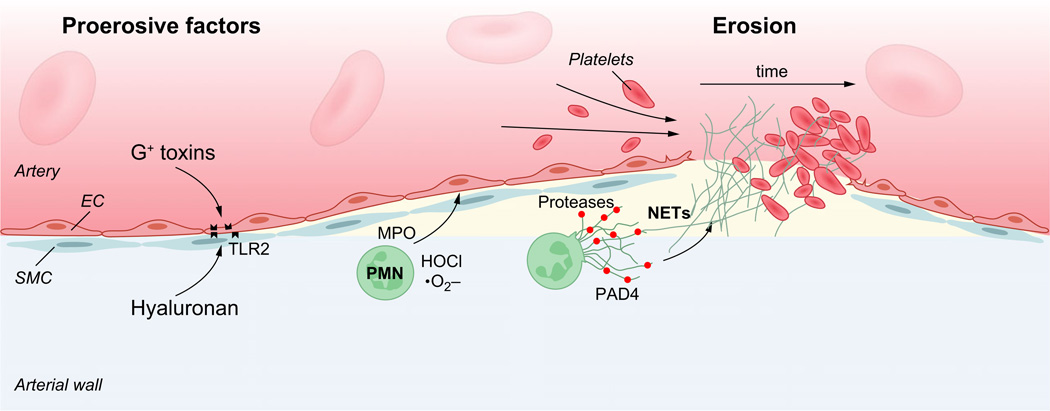
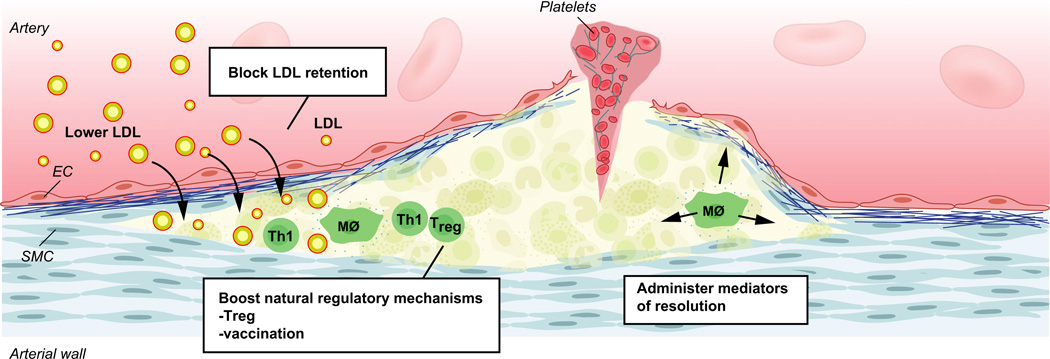
Atherosclerosis is a maladaptive, nonresolving chronic inflammatory disease that occurs at sites of blood flow disturbance. The disease usually remains silent until a breakdown of integrity at the arterial surface triggers the formation of a thrombus. By occluding the lumen, the thrombus or emboli detaching from it elicits ischaemic symptoms that may be life-threatening. Two types of surface damage can cause atherothrombosis: plaque rupture and endothelial erosion. Plaque rupture is thought to be caused by loss of mechanical stability, often due to reduced tensile strength of the collagen cap surrounding the plaque. Therefore, plaques with reduced collagen content are thought to be more vulnerable than those with a thick collagen cap. Endothelial erosion, on the other hand, may occur after injurious insults to the endothelium instigated by metabolic disturbance or immune insults. This review discusses the molecular mechanisms involved in plaque vulnerability and the development of atherothrombosis.
Keywords: atherosclerosis; atherothrombosis; en-dothelial erosion; inflammation; plaque rupture.
© 2015 The Association for the Publication of the Journal of Internal Medicine.
Figures
References
-
- Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. - PubMed
-
- Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of t cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:131–138. - PubMed
-
- Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
