

New Routes to Phylogeography: A Bayesian Structured Coalescent Approximation
- PMID: 26267488
- PMCID: PMC4534465
- DOI: 10.1371/journal.pgen.1005421
New Routes to Phylogeography: A Bayesian Structured Coalescent Approximation
Abstract
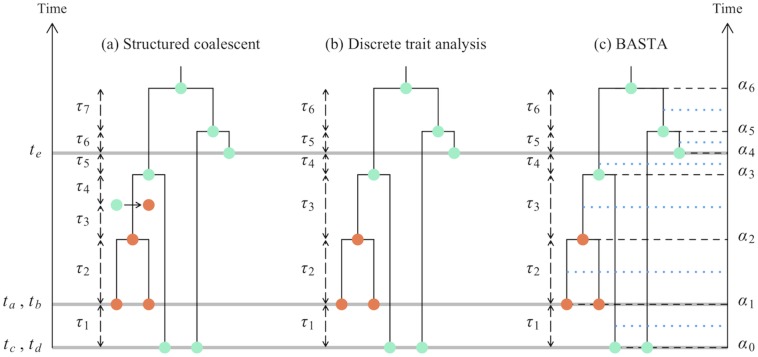
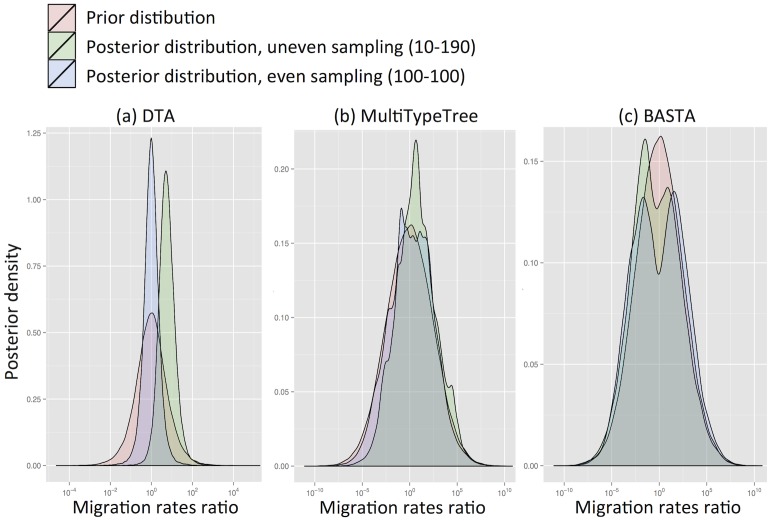
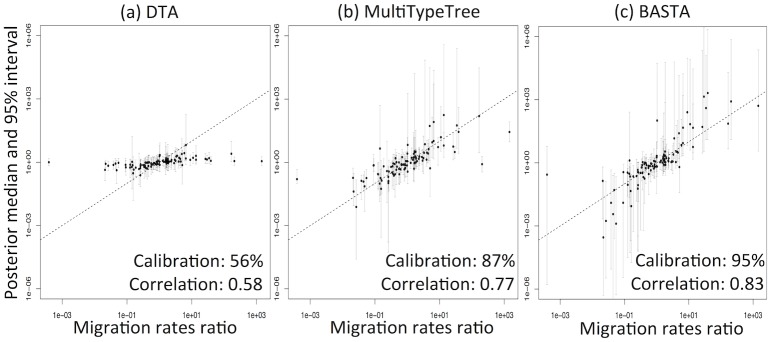
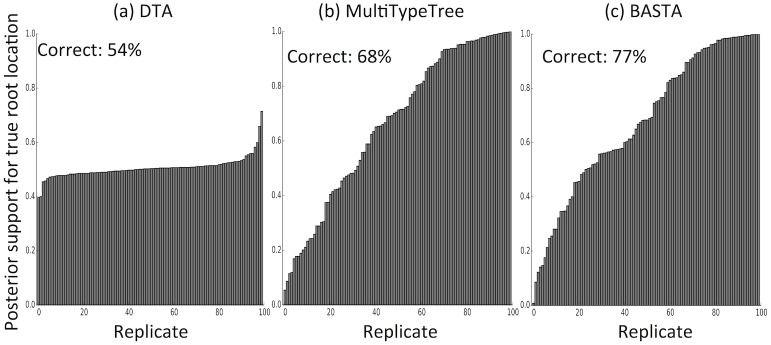
Phylogeographic methods aim to infer migration trends and the history of sampled lineages from genetic data. Applications of phylogeography are broad, and in the context of pathogens include the reconstruction of transmission histories and the origin and emergence of outbreaks. Phylogeographic inference based on bottom-up population genetics models is computationally expensive, and as a result faster alternatives based on the evolution of discrete traits have become popular. In this paper, we show that inference of migration rates and root locations based on discrete trait models is extremely unreliable and sensitive to biased sampling. To address this problem, we introduce BASTA (BAyesian STructured coalescent Approximation), a new approach implemented in BEAST2 that combines the accuracy of methods based on the structured coalescent with the computational efficiency required to handle more than just few populations. We illustrate the potentially severe implications of poor model choice for phylogeographic analyses by investigating the zoonotic transmission of Ebola virus. Whereas the structured coalescent analysis correctly infers that successive human Ebola outbreaks have been seeded by a large unsampled non-human reservoir population, the discrete trait analysis implausibly concludes that undetected human-to-human transmission has allowed the virus to persist over the past four decades. As genomics takes on an increasingly prominent role informing the control and prevention of infectious diseases, it will be vital that phylogeographic inference provides robust insights into transmission history.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
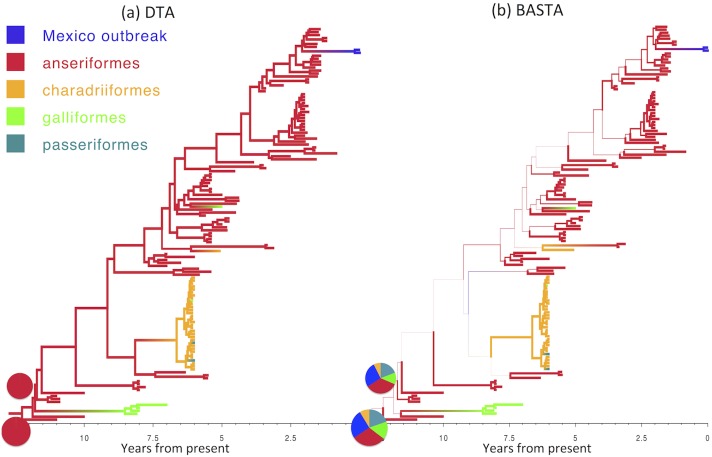
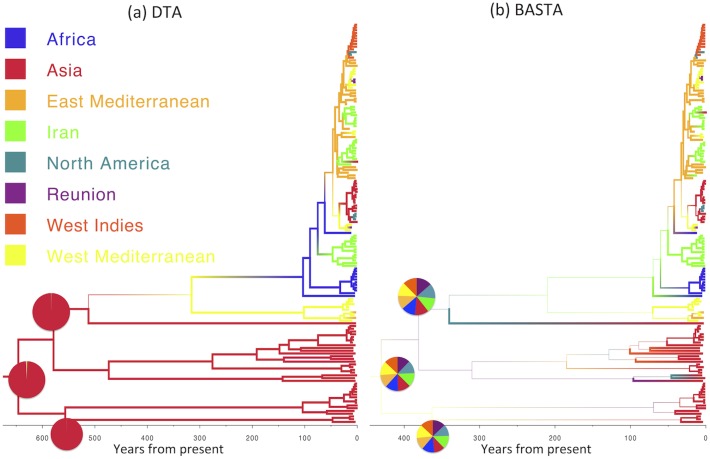
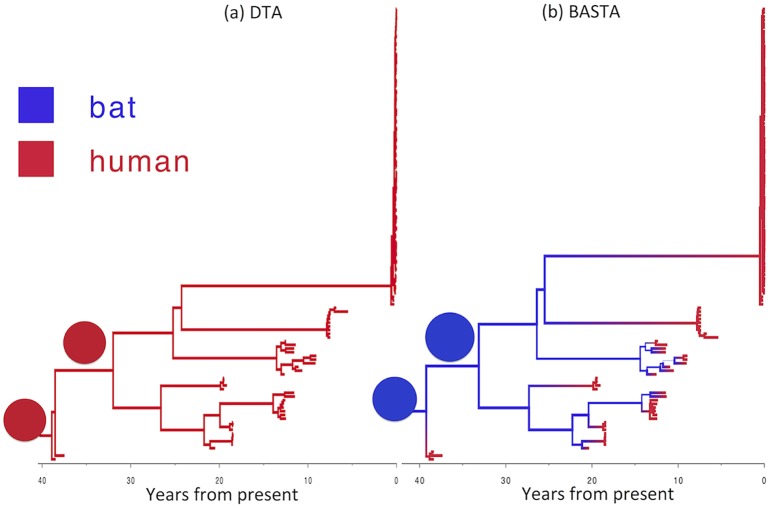
Similar articles
-
Extracting transmission networks from phylogeographic data for epidemic and endemic diseases: Ebola virus in Sierra Leone, 2009 H1N1 pandemic influenza and polio in Nigeria.Int Health. 2015 Mar;7(2):130-8. doi: 10.1093/inthealth/ihv012. Int Health. 2015. PMID: 25733563 Free PMC article.
-
The Structured Coalescent and Its Approximations.Mol Biol Evol. 2017 Nov 1;34(11):2970-2981. doi: 10.1093/molbev/msx186. Mol Biol Evol. 2017. PMID: 28666382 Free PMC article.
-
Going back to the roots: Evaluating Bayesian phylogeographic models with discrete trait uncertainty.Infect Genet Evol. 2020 Nov;85:104501. doi: 10.1016/j.meegid.2020.104501. Epub 2020 Aug 13. Infect Genet Evol. 2020. PMID: 32798768 Free PMC article.
-
Phylogeographic Approaches to Characterize the Emergence of Plant Pathogens.Phytopathology. 2021 Jan;111(1):68-77. doi: 10.1094/PHYTO-07-20-0319-FI. Epub 2020 Nov 21. Phytopathology. 2021. PMID: 33021879 Review.
-
Approaches and challenges to inferring the geographical source of infectious disease outbreaks using genomic data.Lancet Microbe. 2024 Jan;5(1):e81-e92. doi: 10.1016/S2666-5247(23)00296-3. Epub 2023 Nov 30. Lancet Microbe. 2024. PMID: 38042165 Review.
Cited by
-
Aquaculture mediates global transmission of a viral pathogen to wild salmon.Sci Adv. 2021 May 26;7(22):eabe2592. doi: 10.1126/sciadv.abe2592. Print 2021 May. Sci Adv. 2021. PMID: 34039598 Free PMC article.
-
Distinguishing imported cases from locally acquired cases within a geographically limited genomic sample of an infectious disease.Bioinformatics. 2023 Jan 1;39(1):btac761. doi: 10.1093/bioinformatics/btac761. Bioinformatics. 2023. PMID: 36440957 Free PMC article.
-
Accommodating individual travel history, global mobility, and unsampled diversity in phylogeography: a SARS-CoV-2 case study.bioRxiv [Preprint]. 2020 Jun 23:2020.06.22.165464. doi: 10.1101/2020.06.22.165464. bioRxiv. 2020. Update in: Nat Commun. 2020 Oct 9;11(1):5110. doi: 10.1038/s41467-020-18877-9. PMID: 32596695 Free PMC article. Updated. Preprint.
-
MASCOT: parameter and state inference under the marginal structured coalescent approximation.Bioinformatics. 2018 Nov 15;34(22):3843-3848. doi: 10.1093/bioinformatics/bty406. Bioinformatics. 2018. PMID: 29790921 Free PMC article.
-
Biased phylodynamic inferences from analysing clusters of viral sequences.Virus Evol. 2017 Aug 3;3(2):vex020. doi: 10.1093/ve/vex020. eCollection 2017 Jul. Virus Evol. 2017. PMID: 28852573 Free PMC article.
References
-
- Hudson RR, et al. (1990) Gene genealogies and the coalescent process. Oxford surveys in evolutionary biology 7: 44.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
