

Extensive shift in placental transcriptome profile in preeclampsia and placental origin of adverse pregnancy outcomes
- PMID: 26268791
- PMCID: PMC4542630
- DOI: 10.1038/srep13336
Extensive shift in placental transcriptome profile in preeclampsia and placental origin of adverse pregnancy outcomes
Abstract
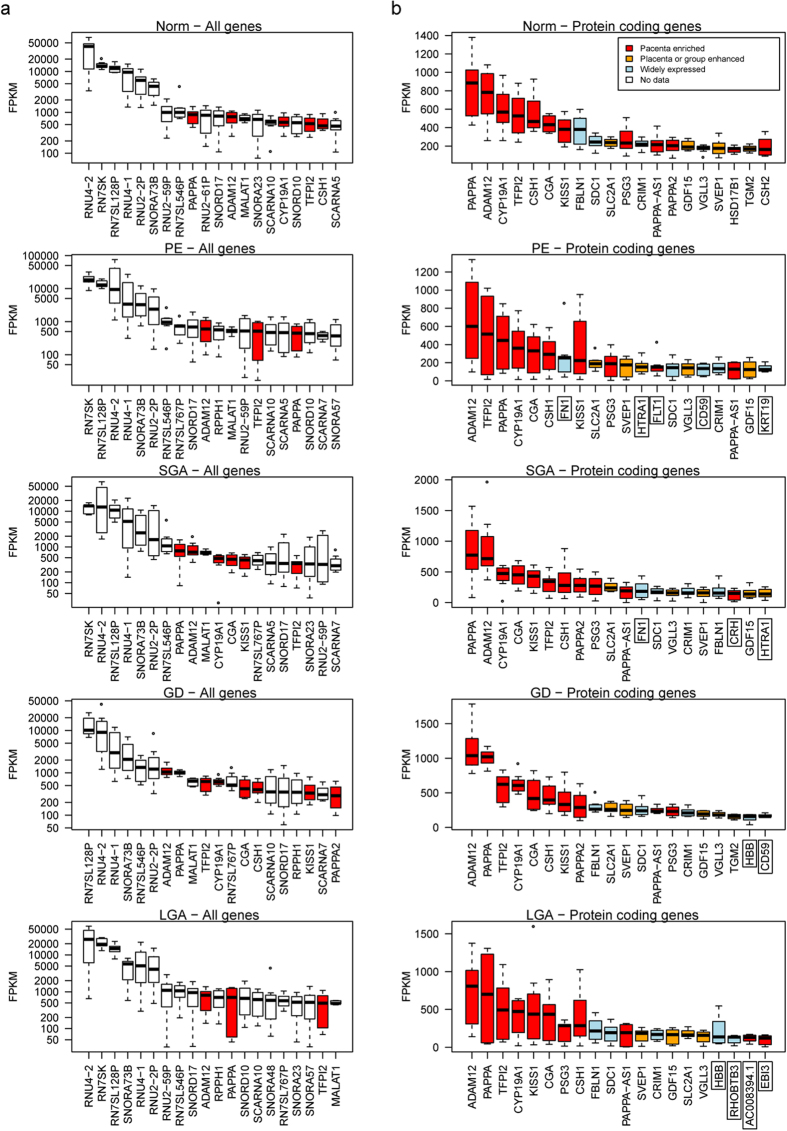
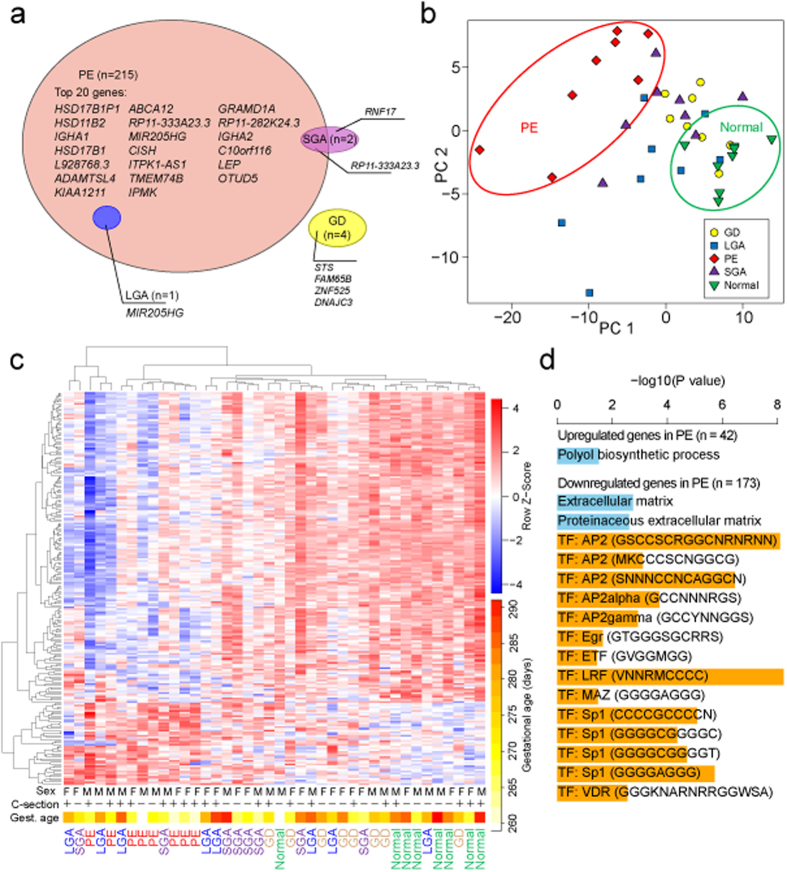
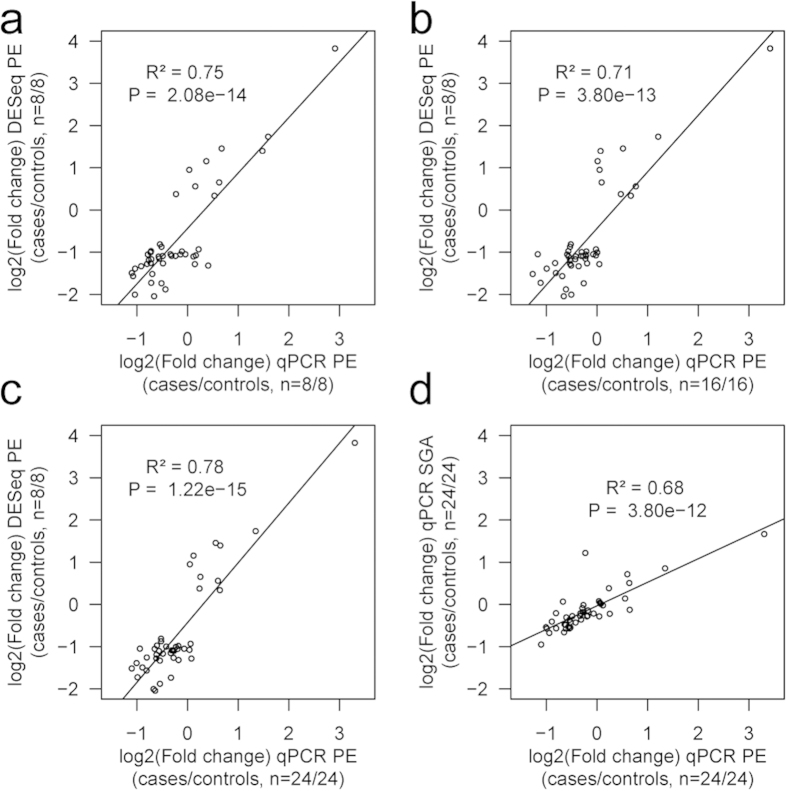
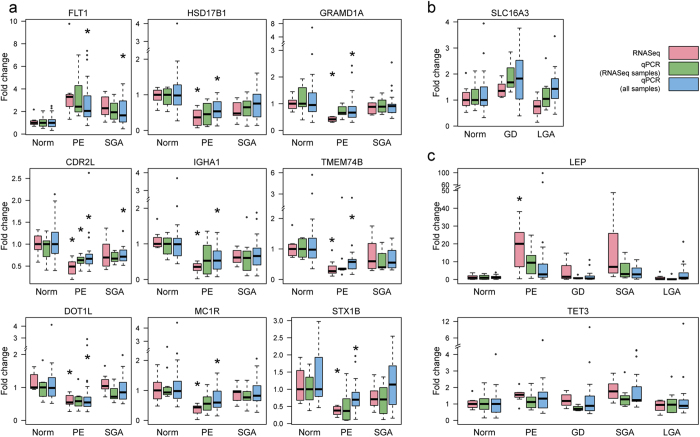
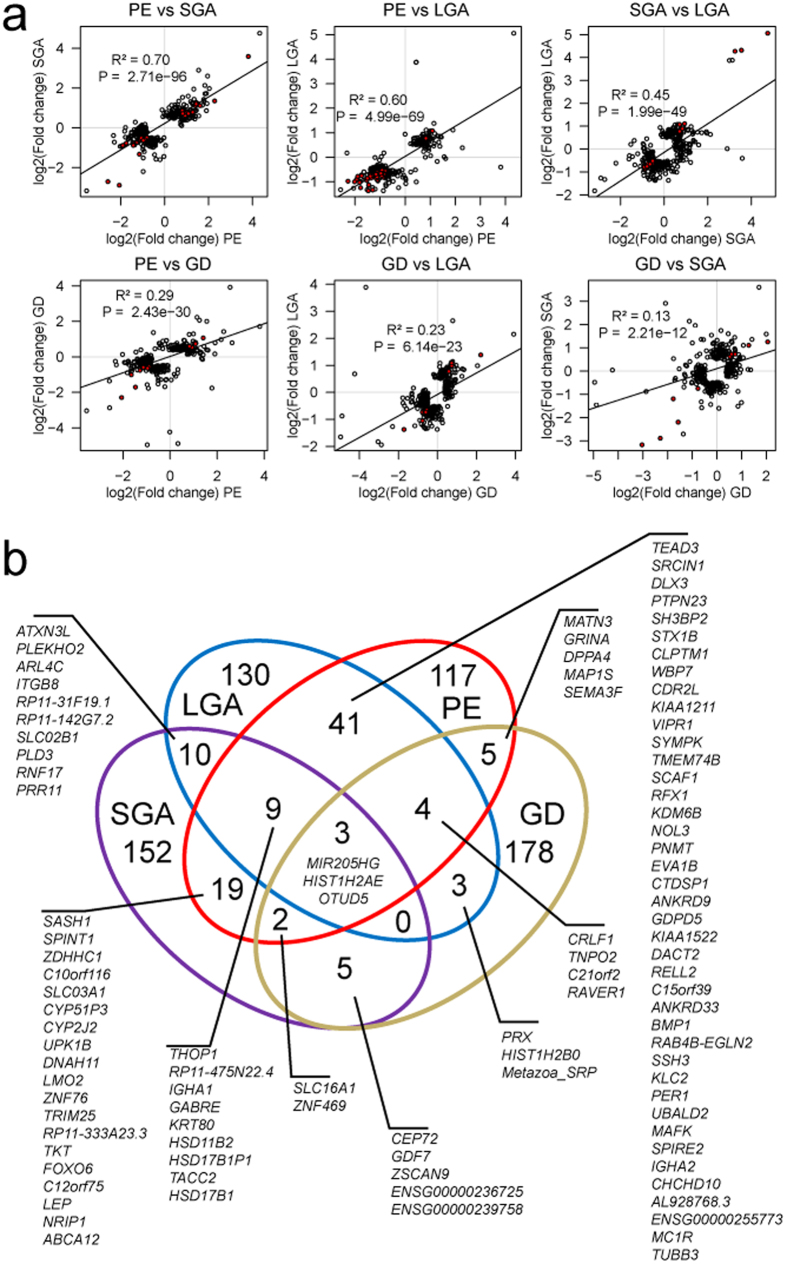
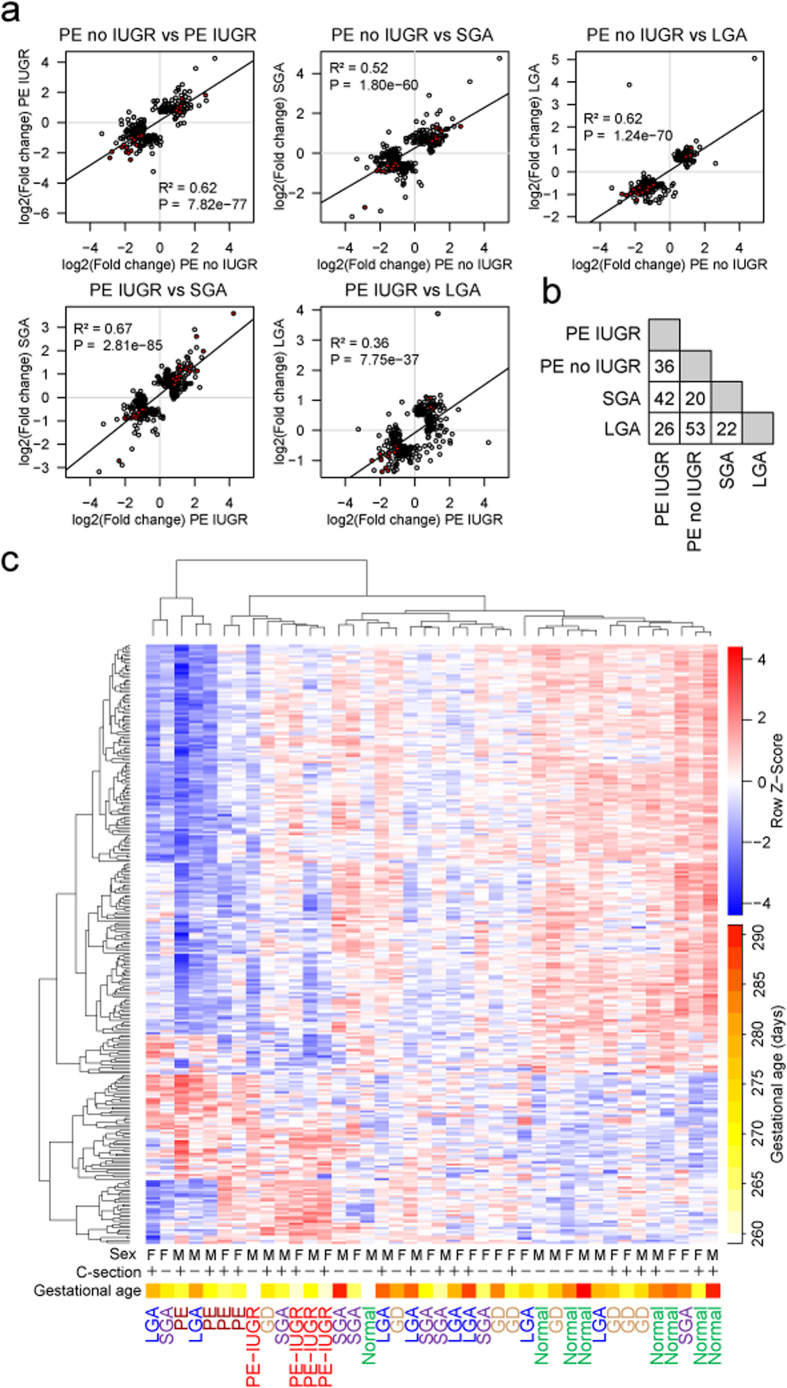
One in five pregnant women suffer from gestational complications, prevalently driven by placental malfunction. Using RNASeq, we analyzed differential placental gene expression in cases of normal gestation, late-onset preeclampsia (LO-PE), gestational diabetes (GD) and pregnancies ending with the birth of small-for-gestational-age (SGA) or large-for-gestational-age (LGA) newborns (n = 8/group). In all groups, the highest expression was detected for small noncoding RNAs and genes specifically implicated in placental function and hormonal regulation. The transcriptome of LO-PE placentas was clearly distinct, showing statistically significant (after FDR) expressional disturbances for hundreds of genes. Taqman RT-qPCR validation of 45 genes in an extended sample (n = 24/group) provided concordant results. A limited number of transcription factors including LRF, SP1 and AP2 were identified as possible drivers of these changes. Notable differences were detected in differential expression signatures of LO-PE subtypes defined by the presence or absence of intrauterine growth restriction (IUGR). LO-PE with IUGR showed higher correlation with SGA and LO-PE without IUGR with LGA placentas. Whereas changes in placental transcriptome in SGA, LGA and GD cases were less prominent, the overall profiles of expressional disturbances overlapped among pregnancy complications providing support to shared placental responses. The dataset represent a rich catalogue for potential biomarkers and therapeutic targets.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Kurt, B. Graham, J. & Burton R. N. B. in Pathol. Hum. Placenta (Springer-Verlag Berlin and Heidelberg GmbH & Co, 2012).
-
- Struwe E. et al.. Microarray analysis of placental tissue in intrauterine growth restriction. Clin. Endocrinol. (Oxf). 72, 241–247 (2010). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
