

The Genome Sequence of Saccharomyces eubayanus and the Domestication of Lager-Brewing Yeasts
- PMID: 26269586
- PMCID: PMC4651232
- DOI: 10.1093/molbev/msv168
The Genome Sequence of Saccharomyces eubayanus and the Domestication of Lager-Brewing Yeasts
Abstract
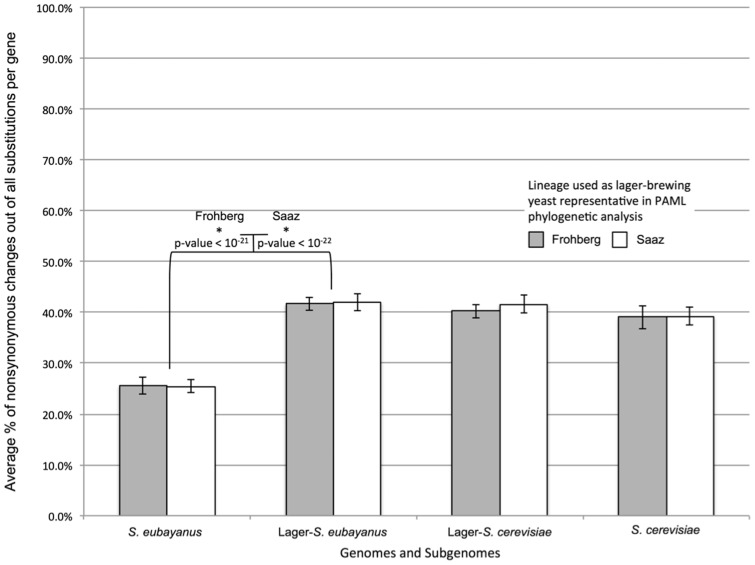
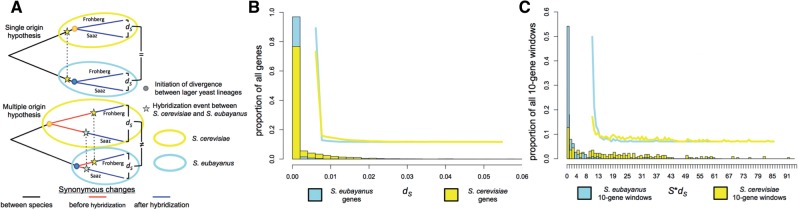
The dramatic phenotypic changes that occur in organisms during domestication leave indelible imprints on their genomes. Although many domesticated plants and animals have been systematically compared with their wild genetic stocks, the molecular and genomic processes underlying fungal domestication have received less attention. Here, we present a nearly complete genome assembly for the recently described yeast species Saccharomyces eubayanus and compare it to the genomes of multiple domesticated alloploid hybrids of S. eubayanus × S. cerevisiae (S. pastorianus syn. S. carlsbergensis), which are used to brew lager-style beers. We find that the S. eubayanus subgenomes of lager-brewing yeasts have experienced increased rates of evolution since hybridization, and that certain genes involved in metabolism may have been particularly affected. Interestingly, the S. eubayanus subgenome underwent an especially strong shift in selection regimes, consistent with more extensive domestication of the S. cerevisiae parent prior to hybridization. In contrast to recent proposals that lager-brewing yeasts were domesticated following a single hybridization event, the radically different neutral site divergences between the subgenomes of the two major lager yeast lineages strongly favor at least two independent origins for the S. cerevisiae × S. eubayanus hybrids that brew lager beers. Our findings demonstrate how this industrially important hybrid has been domesticated along similar evolutionary trajectories on multiple occasions.
Keywords: Saccharomyces eubayanus; domestication; genome assembly; hybridization; lager brewing.
© The Author 2015. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Bing J, Han PJ, Liu WQ, Wang QM, Bai FY. 2014. Evidence for a Far East Asian origin of lager beer yeast. Curr Biol. 24:R380–R381. - PubMed
Publication types
MeSH terms
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
