

MEK plus PI3K/mTORC1/2 Therapeutic Efficacy Is Impacted by TP53 Mutation in Preclinical Models of Colorectal Cancer
- PMID: 26272063
- PMCID: PMC5087596
- DOI: 10.1158/1078-0432.CCR-14-3091
MEK plus PI3K/mTORC1/2 Therapeutic Efficacy Is Impacted by TP53 Mutation in Preclinical Models of Colorectal Cancer
Erratum in
-
Correction: MEK plus PI3K/mTORC1/2 Therapeutic Efficacy Is Impacted by TP53 Mutation in Preclinical Models of Colorectal Cancer.Clin Cancer Res. 2016 Aug 1;22(15):3982. doi: 10.1158/1078-0432.CCR-16-1206. Clin Cancer Res. 2016. PMID: 27480870 No abstract available.
Abstract
Purpose: PI3K pathway activation occurs in concomitance with RAS/BRAF mutations in colorectal cancer, limiting the sensitivity to targeted therapies. Several clinical studies are being conducted to test the tolerability and clinical activity of dual MEK and PI3K pathway blockade in solid tumors.
Experimental design: In the present study, we explored the efficacy of dual pathway blockade in colorectal cancer preclinical models harboring concomitant activation of the ERK and PI3K pathways. Moreover, we investigated if TP53 mutation affects the response to this therapy.
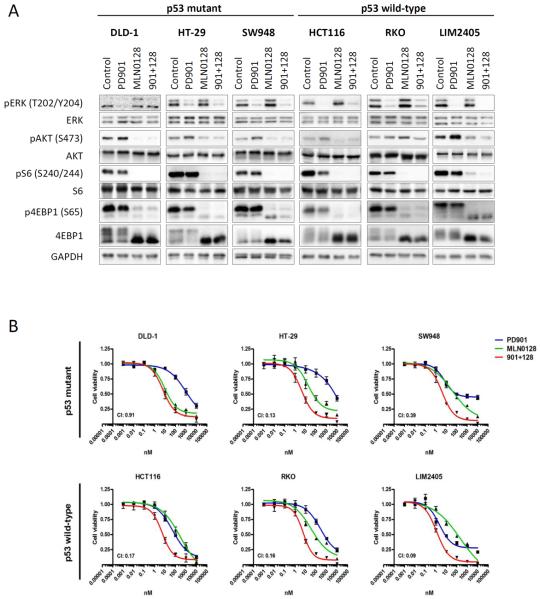
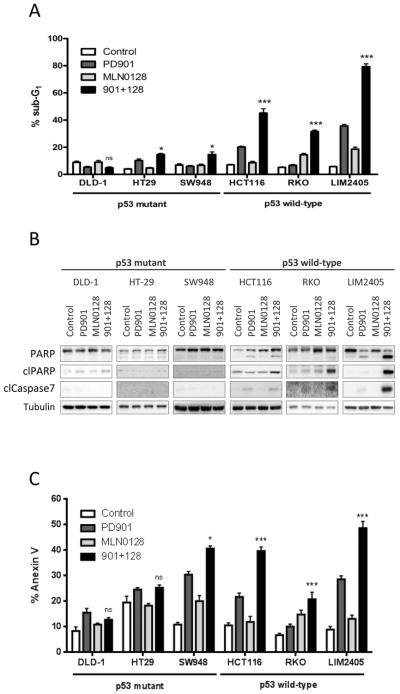
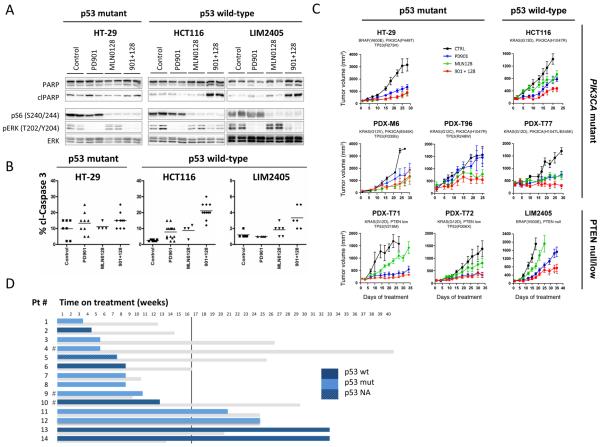
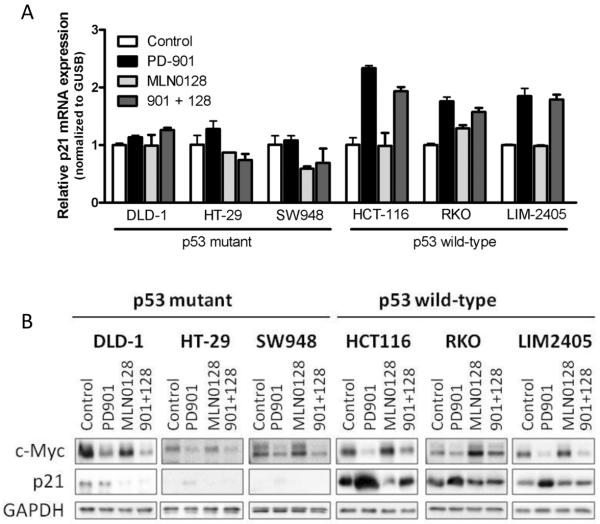
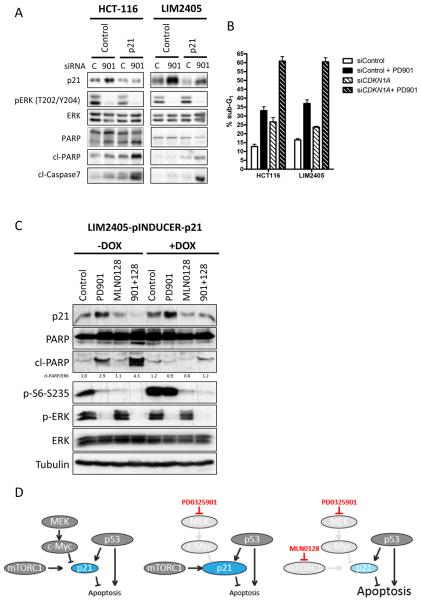
Results: Dual MEK and mTORC1/2 blockade resulted in synergistic antiproliferative effects in cell lines bearing alterations in KRAS/BRAF and PIK3CA/PTEN. Although the on-treatment cell-cycle effects were not affected by the TP53 status, a marked proapoptotic response to therapy was observed exclusively in wild-type TP53 colorectal cancer models. We further interrogated two independent panels of KRAS/BRAF- and PIK3CA/PTEN-altered cell line- and patient-derived tumor xenografts for the antitumor response toward this combination of agents. A combination response that resulted in substantial antitumor activity was exclusively observed among the wild-type TP53 models (two out of five, 40%), but there was no such response across the eight mutant TP53 models (0%). Interestingly, within a cohort of 14 patients with colorectal cancer treated with these agents for their metastatic disease, two patients with long-lasting responses (32 weeks) had TP53 wild-type tumors.
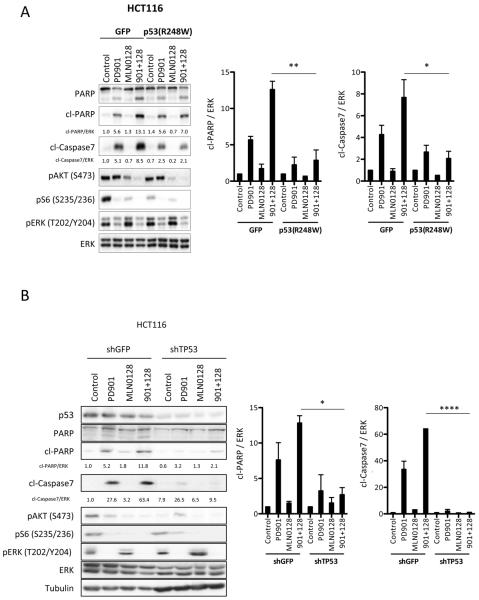
Conclusions: Our data support that, in wild-type TP53 colorectal cancer cells with ERK and PI3K pathway alterations, MEK blockade results in potent p21 induction, preventing apoptosis to occur. In turn, mTORC1/2 inhibition blocks MEK inhibitor-mediated p21 induction, unleashing apoptosis. Clin Cancer Res; 21(24); 5499-510. ©2015 AACR.
©2015 American Association for Cancer Research.
Figures
References
-
- Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26:2013–9. - PubMed
-
- Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–72. - PubMed
-
- Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303–12. - PubMed
-
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67:2643–8. - PubMed
-
- Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69:1851–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
