

Review
doi: 10.1038/nrmicro3521.
Staphylococcal manipulation of host immune responses
Affiliations
- PMID: 26272408
- PMCID: PMC4625792
- DOI: 10.1038/nrmicro3521
Item in Clipboard
Review
Staphylococcal manipulation of host immune responses
Nat Rev Microbiol.
2015 Sep.
Abstract
Staphylococcus aureus, a bacterial commensal of the human nares and skin, is a frequent cause of soft tissue and bloodstream infections. A hallmark of staphylococcal infections is their frequent recurrence, even when treated with antibiotics and surgical intervention, which demonstrates the bacterium's ability to manipulate innate and adaptive immune responses. In this Review, we highlight how S. aureus virulence factors inhibit complement activation, block and destroy phagocytic cells and modify host B cell and T cell responses, and we discuss how these insights might be useful for the development of novel therapies against infections with antibiotic resistant strains such as methicillin-resistant S. aureus.
Figures
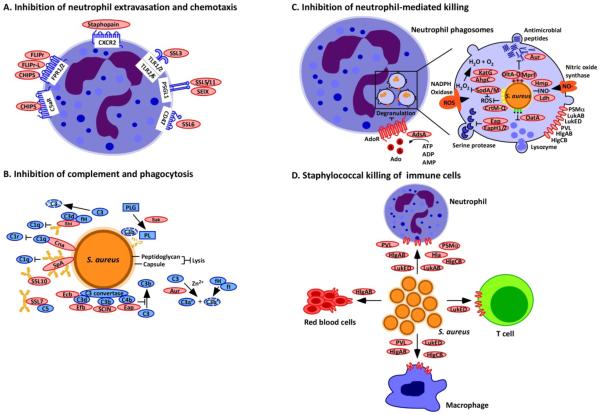
(a) Neutrophil extravasation and chemotaxis is inhibited by Staphylococcus aureus through the secretion of staphylococcal superantigen-like (SSL) molecules. SSL3 inhibits Toll-like receptor heterodimers; SSL5, SSL11 and SElX inhibit PSGL-1 signalling; and SSL6 inhibits the G-protein coupled receptor CD47. Other secreted proteins include chemotaxis inhibitory protein of S. aureus (CHIPS), which inhibits the complement receptor C5aR and the formyl-peptide receptors 1 and 2 (FPR1/2); formyl peptide receptor-like 1 inhibitor (FLIPr) and FLIPr-like (FLIPr-L), which inhibit FPR1/2; and staphopain, which inhibits signalling from the chemokine receptor CXCR2. (b) Complement activation and phagocytosis of staphylococci are blocked through the secretion of inhibitory factors to interfere with opsonization. Cna blocks the association of complement factor C1q bound to immunoglobulin with complement receptor C1r; SpA and Sbi binding to immunoglobulin blocks its association with C1q; Sbi, SpA, SSL7 and SSL10 sequester immunoglobulins to block their abilitiy to promote complement activation; Sbi (when associated with the host factors C3d and fH) and SSL7 also inactivate the complement factors C3 and C5, respectively; Sak associates with plasminogen (PLG) and activates the zymogen to cleave complement factor C3b and immunoglobulin; Efb, Ecb, Eap and SCIN inhibit C3 convertases; and aureolysin cleaves the complement factor C3, which compromises opsonization because the cleavage product C3b is degraded by a complex of the host proteins factor I (fI) and co-factor H (fH). (c)
S. aureus inhibits neutrophil-mediated killing of phagocytosed bacteria by expressing several enzymes and inhibitors. The adenosine-synthesizing enzyme AdsA enables the inhibition of granulation via adenosine receptor (AdoR) signalling; staphyloxanthin, superoxide dismutase (SodA/SodM), the catalase KatG and alkylhydroperoxide reductase (AhpC) are antioxidants that reduce oxidative stress caused by phagosomal reactive oxygen species (ROS) and H2O2 generation; aureolysin (Aur) cleaves antimicrobial peptides; DltA-D promote D-alanyl esterification of teichoic acids to protect staphylococci from antimicrobial peptides; MprF modifies phosphatidylglycerol with alanine or lysine, another mechanism to protect staphylococci against antimicrobial peptides; Ldh and Hmp inhibit nitrosative stress; Eap, EapH1 and EapH2 inhibit neutrophil serine proteases; and OatA O-acetylates peptidoglycan, which prevents its lysozymal degradation. (d) Secreted β-barrel pore forming toxins (β-PFTs), bind specific receptors on immune cells to impair immune cell functions or promote cell lysis. These β-PFTs include LukED, which binds to neutrophils, T cells and macrophages; HlgAB, which binds to neutrophils, macrophages and red blood cells; HlgCB and PVL, which bind to neutrophils and macrophages; and LukAB and Hla, which bind to neutrophils. Phenol-soluble modulin α (PSMα), which is another factor secreted by S. aureus but not a β-PFT, can also lyse white blood cells.
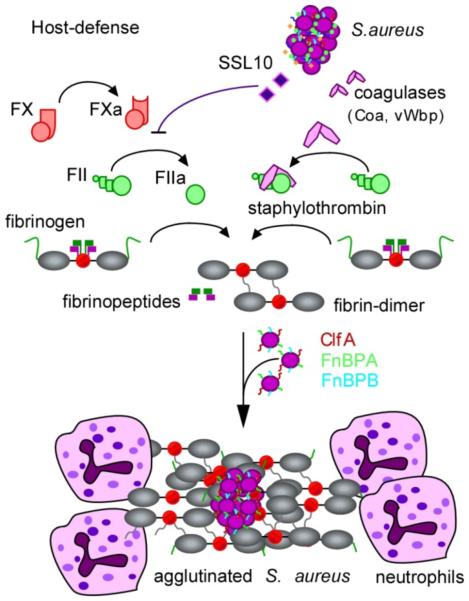
Physiological host defenses immobilize bacteria through the activation of the serine protease zymogens prothrombin (also known as factor II) and factor X (not shown). In the contact activation pathway, surface contact results in the autocleavage of prothrombin (also known as factor II), thereby generating thrombin (also known as factor IIa). The Staphylococcus aureus superantigen-like protein SSL10 inhibits prothrombin autoactivation, whereas the S. aureus coagulases Coa and vWbp convert prothrombin to staphylothrombin. Both thrombin and staphylothrombin cleave fibrinopeptides A and B from fibrinogen to generate fibrin, which self-assembles and polymerizes into cable structures that immobilize bacteria. Thrombin activation results in the activation of additional haemostasis factors that facilitate the simultaneous attraction of phagocytes to immobilized bacteria, which is thus inhibited by SSL10 secretion. However, staphylothrombin cleaves fibrinopeptides from fibrinogen without activation of other haemostasis factors and promotes fibrin polymer assembly on the staphylococcal surface, where it protects the bacterium from neutrophils and phagocytic clearance. Fibrin agglutination on the staphylococcal surface also involves the S. aureus surface proteins ClfA, FnbpB and FnbpB, which bind to the fibrinogen γ-chains.
(a)
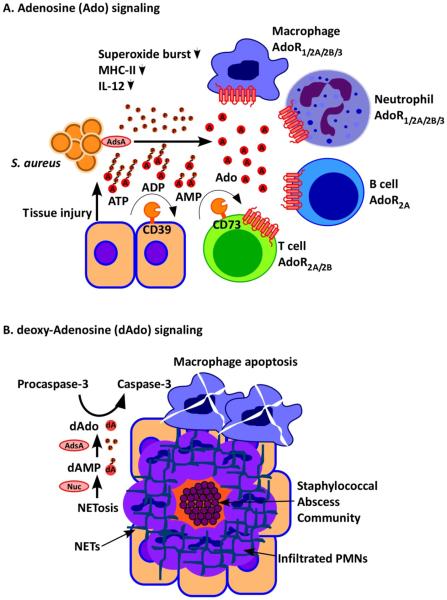
Staphylococcus aureus infection and its associated inflammatory damage promote the release of ATP, which is converted by adenosine synthase A (AdsA) into the immune suppressive signalling molecule adenosine (A). Adenosine inhibits activation of B cells, T cells, macrophages and dendritic cells via adenosine receptor (AdoR) signalling by acting on four different receptors (AdoR1/2A/2B/3). Under physiological conditions, CD39 and CD73 generate adenosine signals to limit inflammatory responses; CD39 and CD73 are also responsible for the adenosine halo surrounding immune cells and for immune suppressive states involving regulatory T cells (T cells expressing the Foxp3+ marker protein (not shown)). (b)
S. aureus induced NETosis of infiltrating neutrophils leads to nuclease-mediated degradation of the DNA fibres that are the major components of neutrophil extracellular traps (NETs) and AdsA-mediated conversion of 5’-monophosphate-deoxyadenosine into deoxyadenosine, which promotes autocleavage of the apoptosis factor pro-capsase 3 to caspase 3. Caspase 3 induces macrophage death, thereby protecting S. aureus against professional phagocytes.
(a)
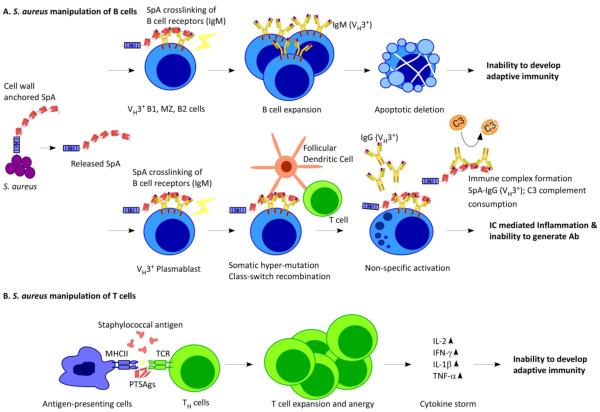
Staphylococcus aureus releases SpA into host tissues, where it binds to and crosslinks VH3 clan B cell receptors. In B1 cells, marginal zone (MZ) B cells and B2 cells, SpA crosslinking is associated with proliferative expansion and apoptotic collapse. The death of these cells impedes the development of adaptive immunity during S. aureus infections. (b) In VH3 plasmablasts, SpA crosslinking promotes somatic hypermutation and class switching from IgM antibodies to IgG antibodies, followed by the secretion of antibodies that are not specific for the S. aureus antigen. (c)
S. aureus secretes T cell superantigen (SAg), which crosslinks major-histocompatibility complex class II antigens (MHCII) on the surface of antigen-presenting cells and T-cell receptors (TCR) on the surface of helper T (TH) cells, triggering T cell expansion and anergy and causing cytokine storms. As a result, T cells specific for S. aureus antigens are not produced.
References
-
- van Belkum A, et al. Co-evolutionary aspects of human colonisation and infection by Staphylococcus aureus. Infect. Genet. Evol. 2009;9:32–47. - PubMed
-
- Kallen AJ, et al. Health care-associated invasive MRSA infections, 2005-2008. JAMA. 2010;304:641–648. - PubMed
-
- Spaan AN, Surewaard BGJ, Nijland R, van Strijp JAG. Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu. Rev. Microbiol. 2013;67:629–650. This excellent review summarizes the molecular events that occur during encounters between neutrophils and staphylococci. - PubMed
-
- Curnutte JT, Whitten DM, Babior BM. Defective superoxide production by granulocytes from patients with chronic granulomatous disease. N. Engl. J. Med. 1974;290:593–597. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
