

Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus
- PMID: 26276630
- PMCID: PMC4537774
- DOI: 10.1016/j.cell.2015.07.020
Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus
Abstract
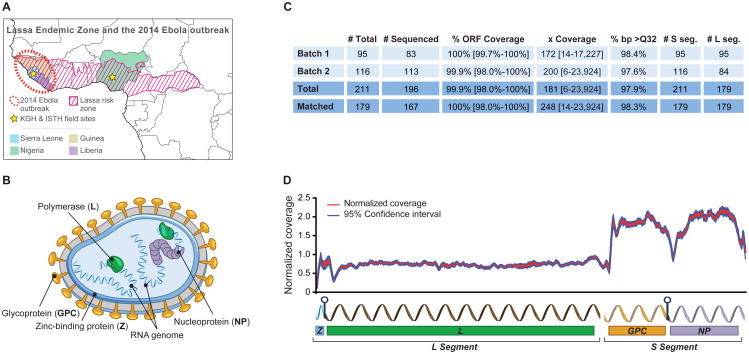
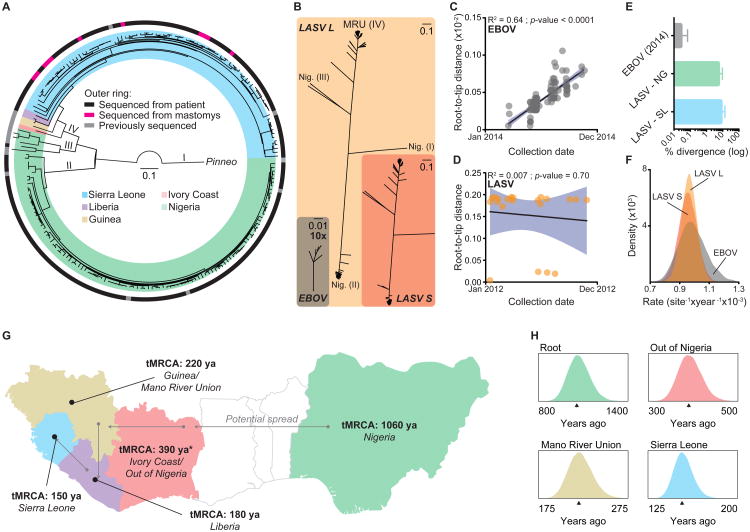
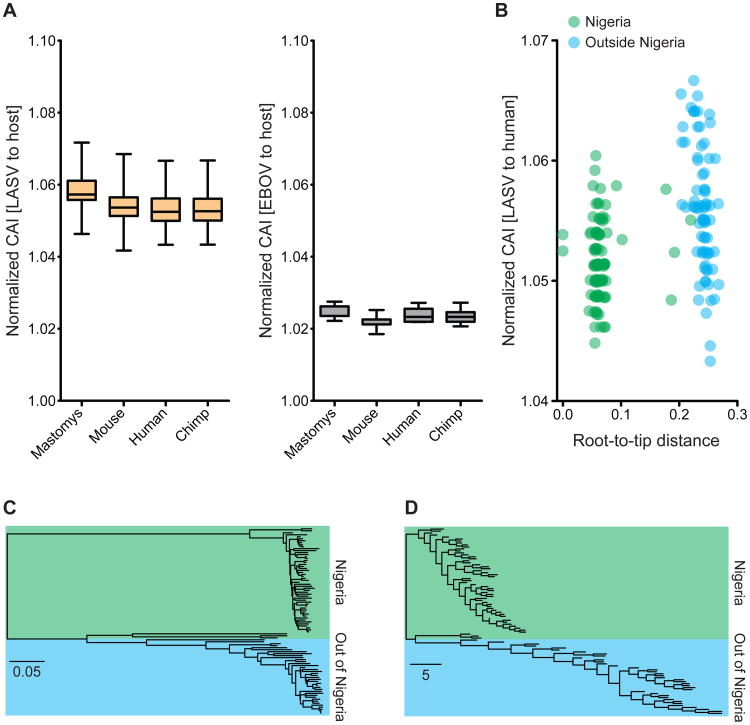
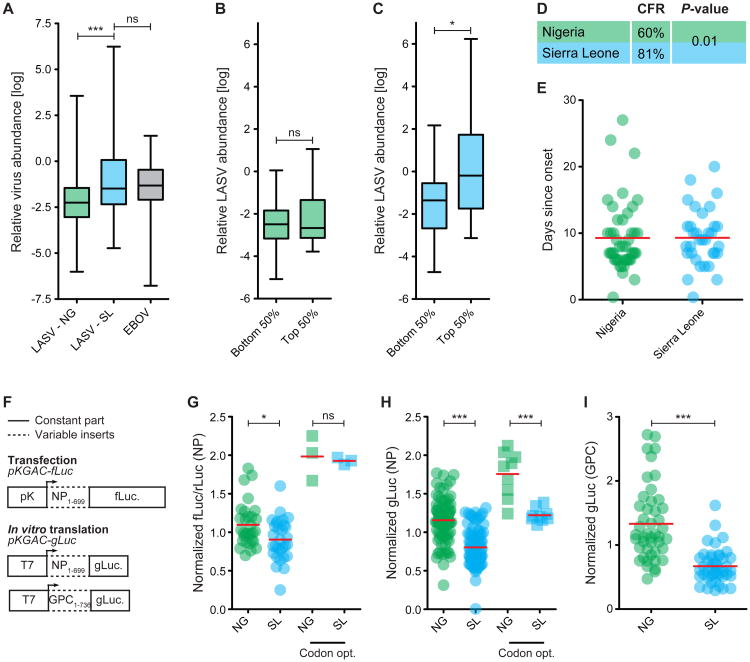
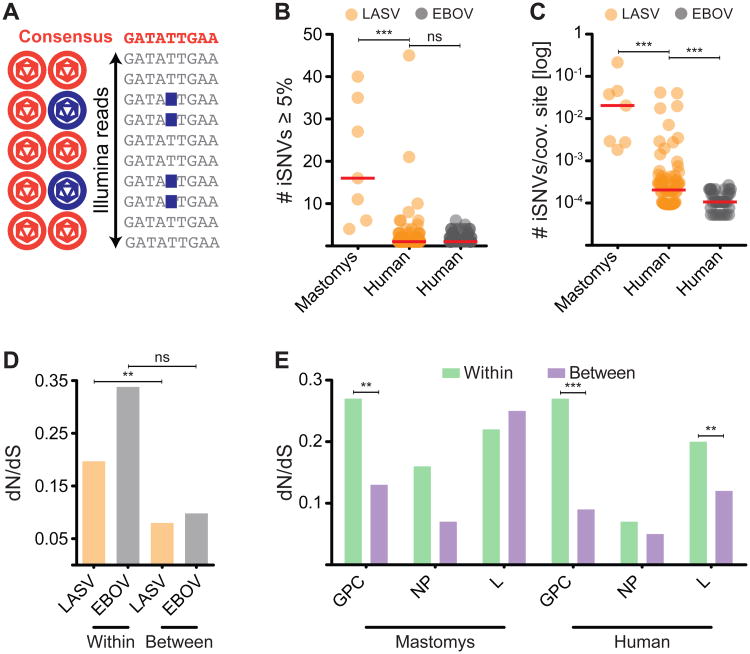
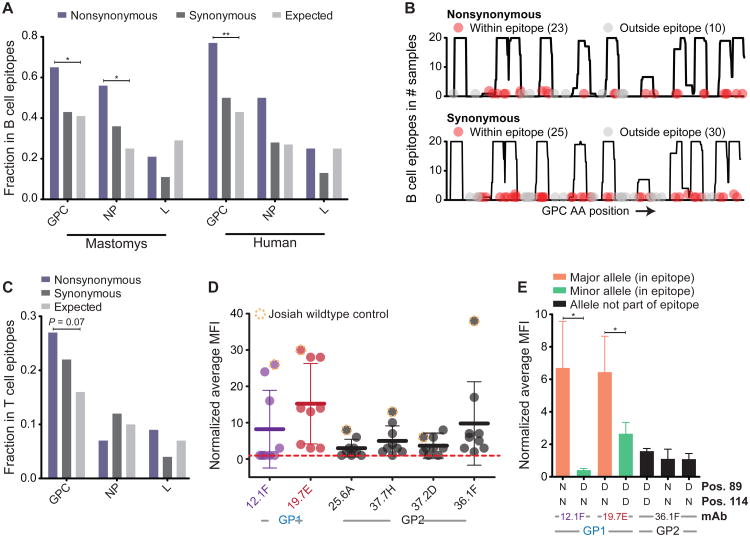
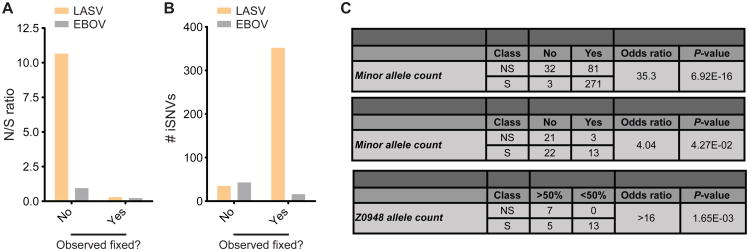
The 2013-2015 West African epidemic of Ebola virus disease (EVD) reminds us of how little is known about biosafety level 4 viruses. Like Ebola virus, Lassa virus (LASV) can cause hemorrhagic fever with high case fatality rates. We generated a genomic catalog of almost 200 LASV sequences from clinical and rodent reservoir samples. We show that whereas the 2013-2015 EVD epidemic is fueled by human-to-human transmissions, LASV infections mainly result from reservoir-to-human infections. We elucidated the spread of LASV across West Africa and show that this migration was accompanied by changes in LASV genome abundance, fatality rates, codon adaptation, and translational efficiency. By investigating intrahost evolution, we found that mutations accumulate in epitopes of viral surface proteins, suggesting selection for immune escape. This catalog will serve as a foundation for the development of vaccines and diagnostics. VIDEO ABSTRACT.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Viral evolution: The history of Lassa virus.Nat Rev Microbiol. 2015 Oct;13(10):600. doi: 10.1038/nrmicro3550. Epub 2015 Aug 24. Nat Rev Microbiol. 2015. PMID: 26299786 No abstract available.
References
-
- Anisimova M, Liberles DA. The quest for natural selection in the age of comparative genomics. Heredity (Edinb) 2007;99:567–579. - PubMed
-
- Baize S, Pannetier D, Oestereich L, Rieger T, Koivogui L, Magassouba N, Soropogui B, Sow MS, Keïta S, De C Hilde, Tiffany A, Dominguez G, Loua M, Traoré A, Kolié M, Malano ER, Heleze E, Bocquin A, Mély S, Raoul H, Caro V, Cadar D, Gabriel M, Pahlmann M, Tappe D, Schmidt-Chanasit J, Impouma B, Diallo AK, Formenty P, Van H Michel, Günther S. Emergence of Zaire Ebola Virus Disease in Guinea —Preliminary Report. N Engl J Med. 2014 140416140039002. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- R01 AI114855/AI/NIAID NIH HHS/United States
- HHSN272201000022C/PHS HHS/United States
- HHSN272200900018C/AI/NIAID NIH HHS/United States
- 1DP2OD006514-01/OD/NIH HHS/United States
- U19 AI109762/AI/NIAID NIH HHS/United States
- K99 HG008179/HG/NHGRI NIH HHS/United States
- HHSN272200900018C/PHS HHS/United States
- HHSN272201000022C/AI/NIAID NIH HHS/United States
- U19AI110818/AI/NIAID NIH HHS/United States
- HHSN272200900049C/AI/NIAID NIH HHS/United States
- DP2 OD006514/OD/NIH HHS/United States
- U19 AI110818/AI/NIAID NIH HHS/United States
- HHSN272200900049C/PHS HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
