

Oncogenes strike a balance between cellular growth and homeostasis
- PMID: 26277544
- PMCID: PMC4662909
- DOI: 10.1016/j.semcdb.2015.08.005
Oncogenes strike a balance between cellular growth and homeostasis
Abstract
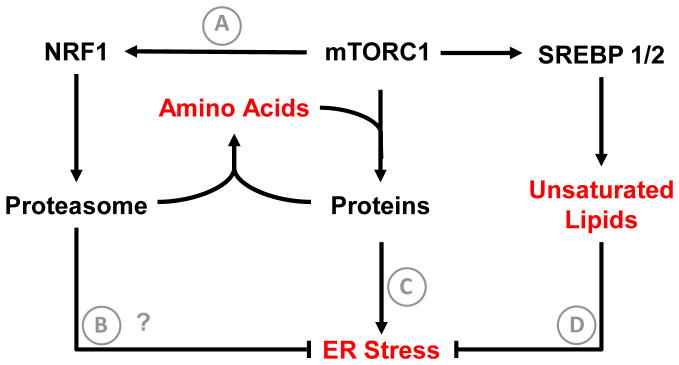
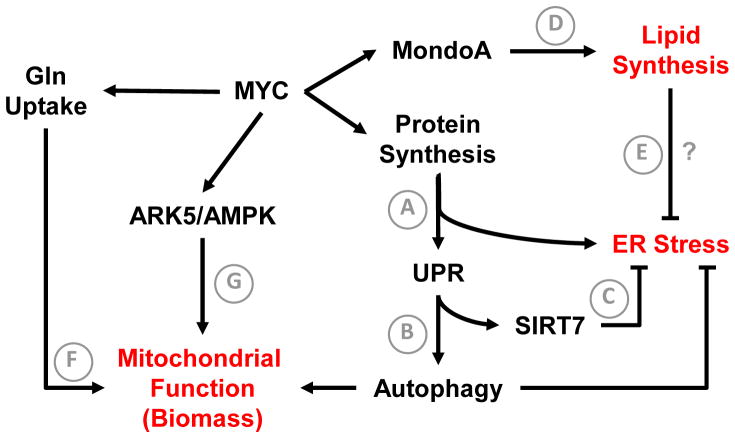
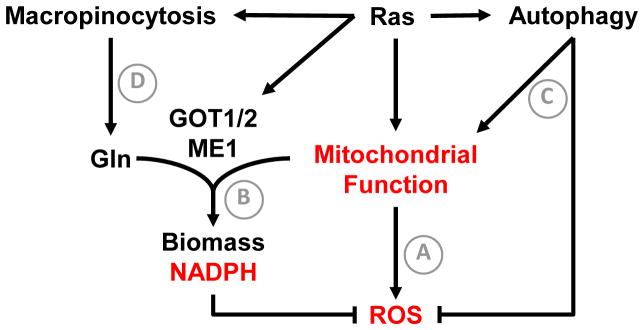
Altered tumor cell metabolism is now firmly established as a hallmark of human cancer. Downstream of oncogenic events, metabolism is re-wired to support cellular energetics and supply the building blocks for biomass. Rapid, uncontrolled proliferation results in tumor growth beyond the reach of existing vasculature and triggers cellular adaptations to overcome limiting nutrient and oxygen delivery. However, oncogenic activation and metabolic re-programming also elicit cell intrinsic stresses, independent of the tumor microenvironment. To ensure metabolic robustness and stress resistance, pro-growth signals downstream of oncogene activation or tumor suppressor loss simultaneously activate homeostatic processes. Here, we summarize recent literature describing the adaptive mechanisms co-opted by common oncogenes, including mTOR, MYC, and RAS. Recurrent themes in our review include: (1) coordination of oncogene-induced changes in protein and lipid metabolism to sustain endoplasmic reticulum homeostasis, (2) maintenance of mitochondrial functional capacity to support anabolic metabolism, (3) adaptations to sustain intracellular metabolite concentrations required for growth, and (4) prevention of oxidative stress. We also include a discussion of the hypoxia inducible factors (HIFs) and the AMP-dependent protein kinase (AMPK)--stress sensors that are co-opted to support tumor growth. Ultimately, an understanding of the adaptations required downstream of specific oncogenes could reveal targetable metabolic vulnerabilities.
Keywords: AMPK; Autophagy; Cancer metabolism; ER stress; Hypoxia inducible factors; Stress response.
Copyright © 2015 Elsevier Ltd. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
