

Unfolded Protein Response and PERK Kinase as a New Therapeutic Target in the Pathogenesis of Alzheimer's Disease
- PMID: 26282939
- PMCID: PMC5010237
- DOI: 10.2174/0929867322666150818104254
Unfolded Protein Response and PERK Kinase as a New Therapeutic Target in the Pathogenesis of Alzheimer's Disease
Abstract
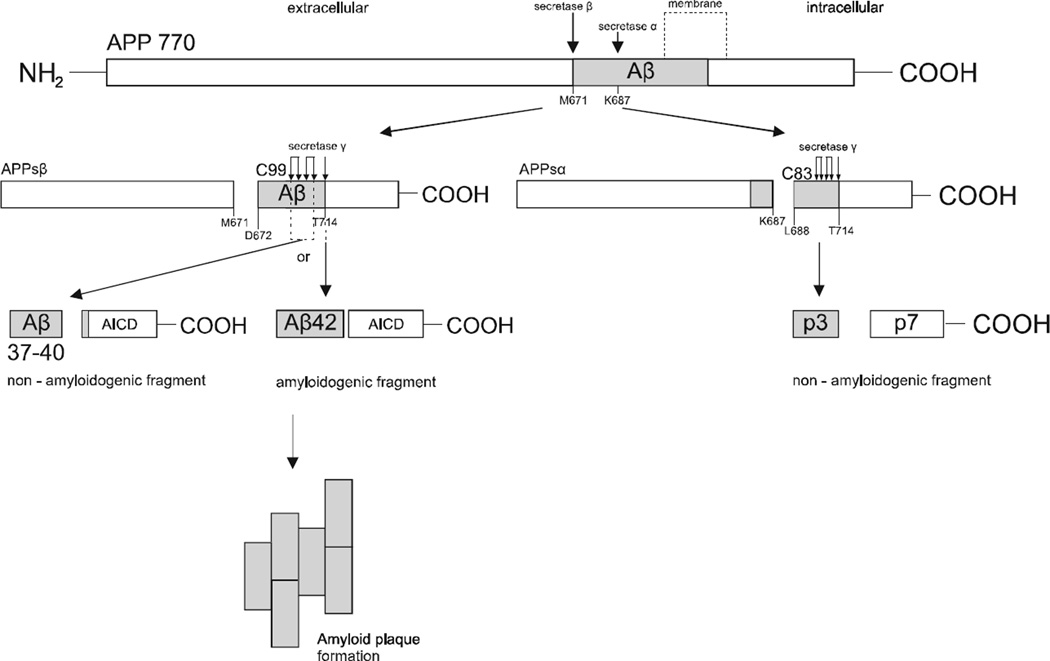
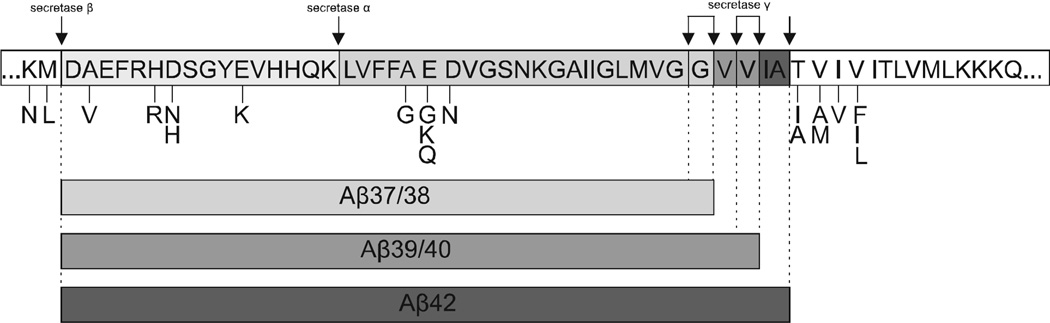
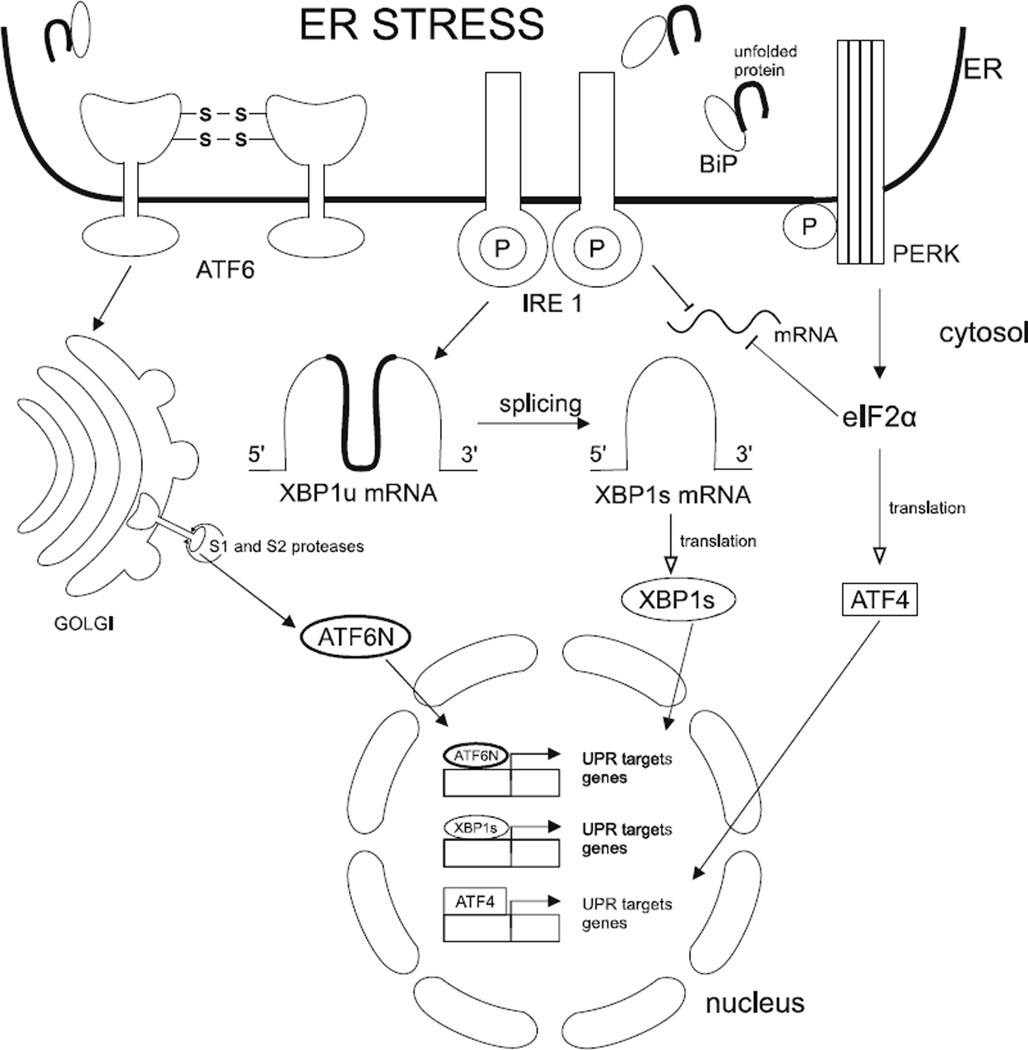
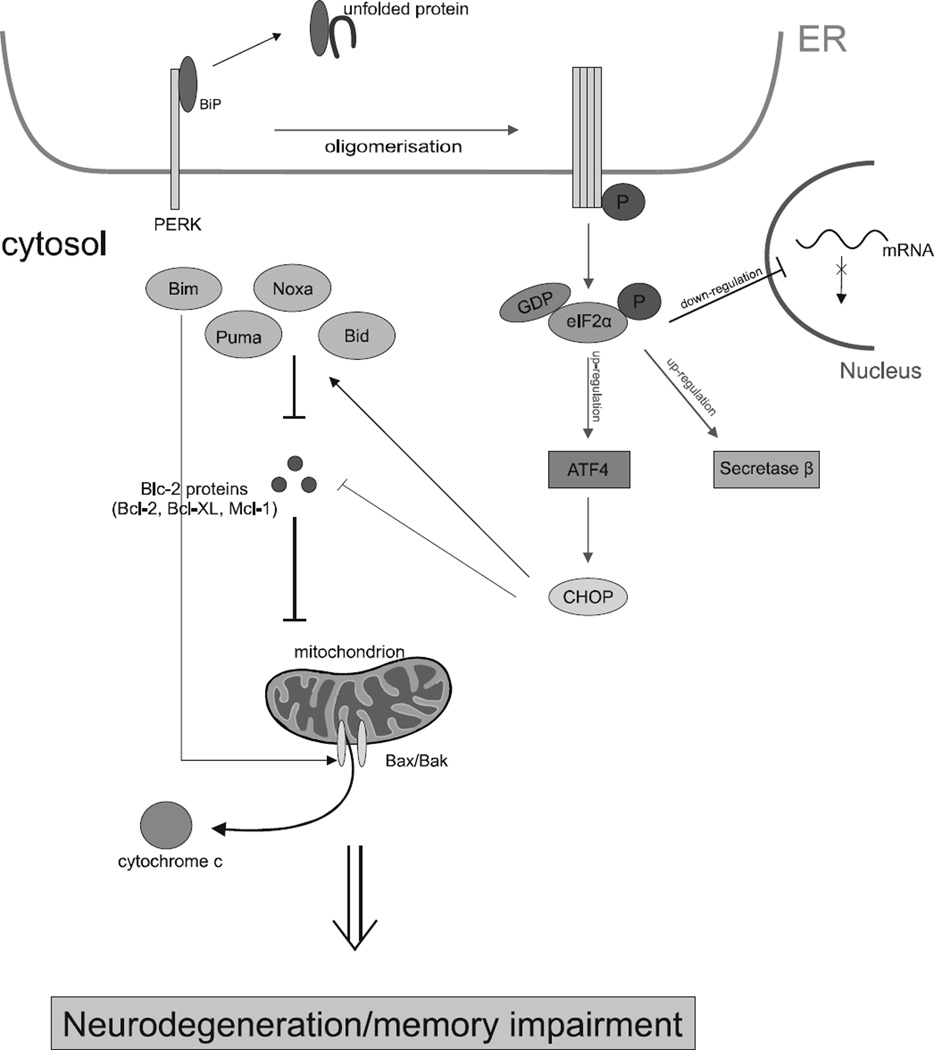
Recent evidence suggests that the development of Alzheimer's disease (AD) and related cognitive loss is due to mutations in the Amyloid Precursor Protein (APP) gene on chromosome 21 and increased activation of eukaryotic translation initiation factor-2α (eIF2α) phosphorylation. The high level of misfolded and unfolded proteins loading in Endoplasmic Reticulum (ER) lumen triggers ER stress and as a result Unfolded Protein Response (UPR) pathways are activated. Stress-dependent activation of the protein kinase RNA-like endoplasmic reticulum kinase (PERK) leads to the significant elevation of phospho-eIF2α. That attenuates general translation and, on the other hand, promotes the preferential synthesis of Activating Transcription Factor 4 (ATF4) and secretase β (BACE1) - a pivotal enzyme responsible for the initiation of the amyloidogenic pathway resulting in the generation of the amyloid β (Aβ) variant with high ability to form toxic senile plaques in AD brains. Moreover, excessive, long-term stress conditions may contribute to inducing neuronal death by apoptosis as a result of the overactivated expression of pro-apoptotic proteins via ATF4. These findings allow to infer that dysregulated translation, increased expression of BACE1 and ATF4, as a result of eIF2α phosphorylation, may be a major contributor to structural and functional neuronal loss resulting in memory impairment. Thus, blocking PERK-dependent eIF2α phosphorylation through specific, small-molecule PERK branch inhibitors seems to be a potential treatment strategy for AD individuals. That may contribute to the restoration of global translation rates and reduction of expression of ATF4 and BACE1. Hence, the treatment strategy can block accelerated β -amyloidogenesis by reduction in APP cleaving via the BACE1-dependent amyloidogenic pathway.
Conflict of interest statement
The authors confirm that this article content has no conflict of interest.
Figures
References
-
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR. Sustained translational repression by eIF2alpha-P mediates prion neurode-generation. Nature. 2012;485(7399):507–511. - PMC - PubMed
-
- Mayeux R. Epidemiology of neurodegeneration. Ann. Rev. Neurosci. 2003;26:81–104. - PubMed
-
- Blacker D, Tanzi RE. The genetics of Alzheimer disease: current status and future prospects. Arch. Neurol. 1998;55(3):294–296. - PubMed
-
- Philipson O, Lord A, Gumucio A, O’Callaghan P, Lannfelt L, Nilsson LN. Animal models of amyloid-beta-related pathologies in Alzheimer’s disease. FEBS J. 2010;277(6):1389–1409. - PubMed
-
- Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999;65(3):664–670. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
