

Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: overlap between Aicardi-Goutières and Singleton-Merten syndromes
- PMID: 26284909
- PMCID: PMC4745891
- DOI: 10.1111/bjd.14073
Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: overlap between Aicardi-Goutières and Singleton-Merten syndromes
Abstract
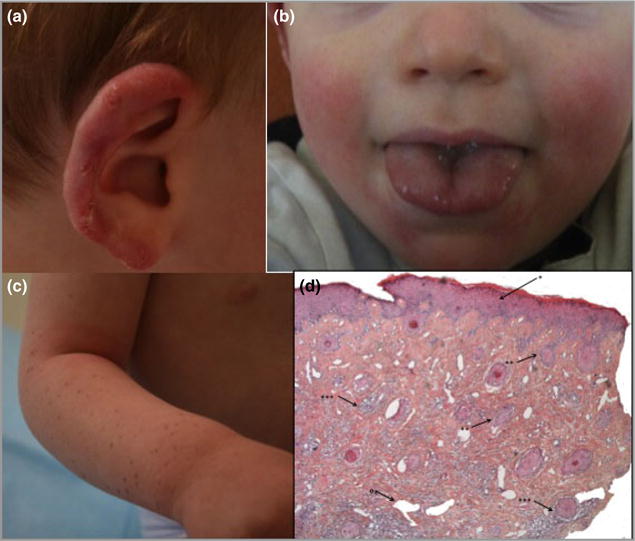
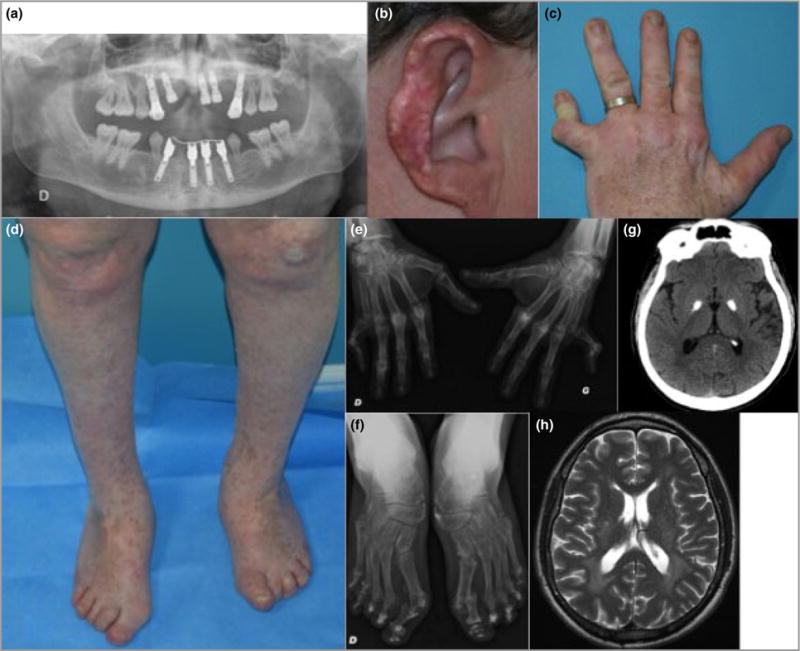
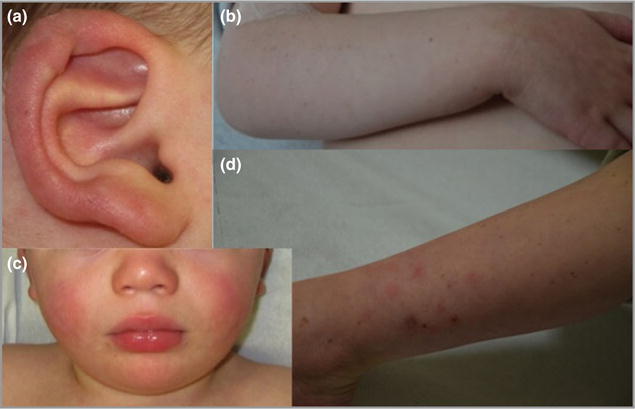
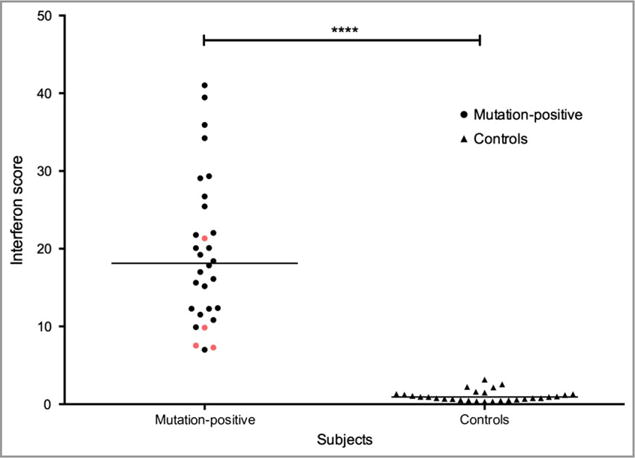
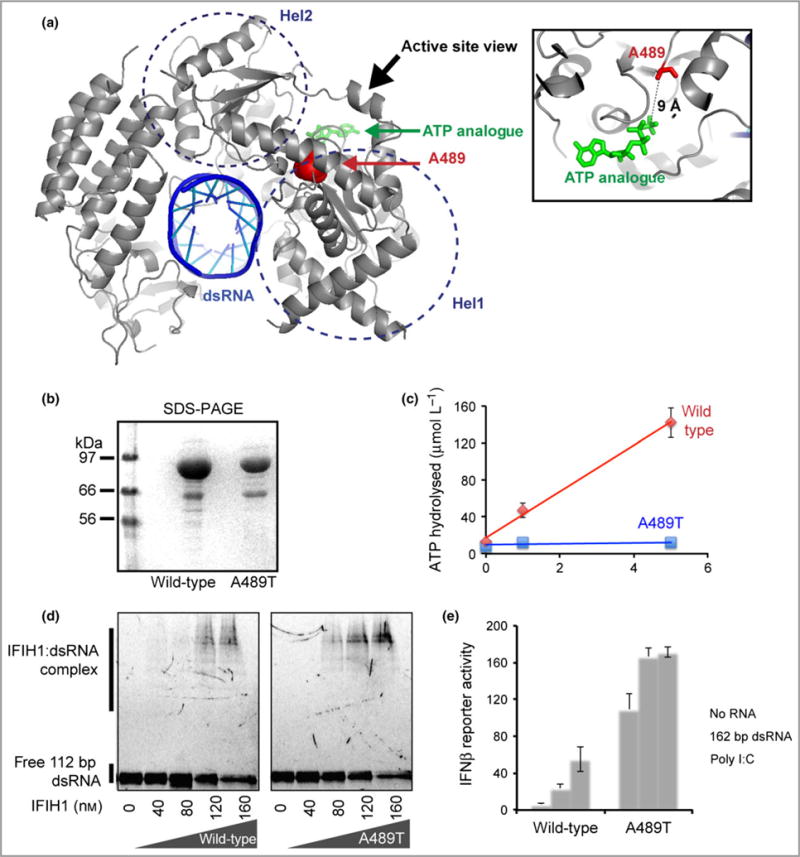
Cutaneous lesions described as chilblain lupus occur in the context of familial chilblain lupus or Aicardi-Goutières syndrome. To date, seven genes related to Aicardi-Goutières syndrome have been described. The most recently described encodes the cytosolic double-stranded RNA receptor IFIH1 (also known as MDA5), a key component of the antiviral type I interferon-mediated innate immune response. Enhanced type I interferon signalling secondary to gain-of-function mutations in IFIH1 can result in a range of neuroinflammatory phenotypes including classical Aicardi-Goutières syndrome. It is of note that none of the patients with a neurological phenotype so far described with mutations in this gene was reported to demonstrate cutaneous involvement. We present a family segregating a heterozygous pathogenic mutation in IFIH1 showing dermatological involvement as a prominent feature, variably associated with neurological disturbance and premature tooth loss. All three affected individuals exhibited increased expression of interferon-stimulated genes in whole blood, and the mutant protein resulted in enhanced interferon signalling in vitro, both in the basal state and following ligand stimulation. Our results further extend the phenotypic spectrum associated with mutations in IFIH1, indicating that the disease can be confined predominantly to the skin, while also highlighting phenotypic overlap with both Aicardi-Goutières syndrome and Singleton-Merten syndrome.
© 2015 British Association of Dermatologists.
Conflict of interest statement
None declared.
Figures
Comment in
-
Chilblain lesions associated with inherited autoimmune disease.Br J Dermatol. 2015 Dec;173(6):1369-70. doi: 10.1111/bjd.14210. Br J Dermatol. 2015. PMID: 26708548 No abstract available.
References
-
- Günther C, Meurer M, Stein A, et al. Familial chilblain lupus – a monogenic form of cutaneous lupus erythematosus due to a heterozygous mutation in TREX1. Dermatology. 2009;219:162–6. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
